

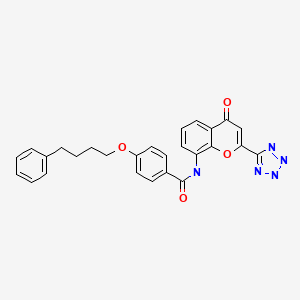

PRANLUKAST
WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ]
Home » 2014 (Page 10)
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
PRANLUKAST
| Launched – 1995 japan |

Pranlukast is a cysteinyl leukotriene receptor-1 antagonist. This drug works similarly to Merck & Co.‘s Singulair (montelukast). It is widely used in Japan.
Medications of this class, which go under a variety of names according to whether one looks at the American, British or European system of nomenclature, have as their primary function the antagonism of bronchospasm caused, principally in asthmatics, by an allergic reaction to accidentally or inadvertently encountered allergens.
Medications of this group are normally used as an adjunct to the standard therapy of inhaled steroids with inhaled long- and/or short-acting beta-agonists. There are several similar medications in the group; all appear to be equally effective.
Pranlukast hydrate is a leukotriene CysLT1 (LTD4) and CysLT2 (LTC4) antagonist first launched in Japan in 1995 as capsules for the oral treatment of bronchial asthma and allergic rhinitis. A dry syrup formulation of pranlukast for the treatment of asthma was approved in Japan in 1999. In April 2011, Ono filed a regulatory application in Japan seeking approval of the compound for the treatment of allergic rhinitis in pediatric patients. In December 2011, approval was obtained for this indication and launch took place immediately.
In terms of clinical development, Ono had been evaluating the drug in phase III for the treatment of sinusitis; however, in 2008 the compound was discontinued for this indication when the compound failed to demostrate the expected efficacy in the phase III studies. In March 2006, Ono discontinued development of the compound for the oral treatment of chronic obstructive pulmonary disease (COPD) based on results which suggested no evidence of efficacy. In 2000, Ono signed a license agreement with Schering-Plough to develop and market pranlukast hydrate in Latin America.
UNII-FR702N558K
4-Oxo-8-[(4-phenylbutoxy)benzoylamino]-2-(tetrazol-5-yl)-4H-1-benzopyran · 1/2 hydrate (common name: pranlukast, hereinafter referred to as “pranlukast” in the specification including the claims) represented by formula:

is a compound having a potential antagonistic action against leucotriene C4(LTC4) and leucotriene D4 (LTD4) and is expected as a treating agent for allergic bronchial or pulmonary diseases, allergic shock, and various allergic inflammatory diseases.

 |
|
| Systematic (IUPAC) name | |
|---|---|
| N-[4-oxo-2-(1H-tetrazol-5-yl)-4H-chromen-7-yl]-4-(4-phenylbutoxy)benzamide | |
| Clinical data | |
| AHFS/Drugs.com | International Drug Names |
| Legal status |
?
|
| Routes | Oral |
| Pharmacokinetic data | |
| Metabolism | Hepatic (mainly CYP3A4)[1] |
| Half-life | 1.5 hours[1] |
| Identifiers | |
| CAS number | 103177-37-3 |
| ATC code | R03DC02 |
| PubChem | CID 4887 |
| DrugBank | DB01411 |
| ChemSpider | 4718 |
| UNII | TB8Z891092 |
| ChEMBL | CHEMBL21333 |
| Chemical data | |
| Formula | C27H23N5O4 |
| Mol. mass | 481.503 g/mol |









 ………………………..
………………………..

Example 1: Synthesis of pranlukast
To 10 g of N-(4-oxo-2-(l-trityl-lH-tetrazol-5-yl)-4H-chromen-8-yl)-4-(4- phenylbutoxy) benzamide (Pharmacostech) was added 100 ml of methanol, and 10 g of a resin pre-treated with hydrochloric acid of pH 2-3 (TRILITE SCR-B gel type,
Mitsubishi Chemical Co.) was added to the reaction mixture, followed by refluxing for
5 hours. The solid components were filtered out from the reaction mixture and washed with 100 ml of methanol. The filter-in solution was subject to vacuum distillation to obtain a solid substance and the solid was dissolved in 50 ml of dimethyl acetamide (DMAC). Afterwards, 200 ml of aqueous solution was added to the DMAC solution and stirred for 1 hour at room temperature. Then, the solid formed was filtered out, dried, and left for 5 hours at room temperature to give 6.32 g (yield:
95%) of the standard compound represented by the following Formula 5: melting point, 231-2330C (decomposed); 1H-NMR (DMSOd6, 300 MHz), δ 1.9 (m, 4H), 2,7 (m,2H), 4.0 (t, 2H), 7.0 (s, 2H), 7.1 (s, IH), 7.2-7.3 (m, 5H), 7.6 (t, IH), 7.9 (t, IH), 8.0
(m, 2H), 8.3 (t, IH), 10.0 (bs, IH).

Example 2: Synthesis of pranlukastOne hundred ml of methanol was added to 10 g of N-(4-oxo-2-(l-trityl-lH- tetrazol-5-yl)-4H-chromen-8-yl)-4-(4-phenylbutoxy) benzamide (Pharmacostech), then 10 g of resin pre-treated with hydrochloric acid of pH 2-3 (TRILITE SCR-10 gel type, Mitsubishi Chemical Co.) was added to the reaction mixture, followed by refluxing for 6 hours. The solid components were filtered out from the reaction mixture and washed with 100 ml of methanol. The filter-in solution was subject to vacuum distillation to obtain a solid substance and the solid was dissolved in 50 ml of dimethyl acetamide (DMAC). Afterwards, 200 ml of aqueous solution was added to the DMAC solution and stirred for 1 hour at room temperature. Then the solid formed was filtered out, dried, and left for 5 hours at room temperature to obtain 6.18 g (yield rate: 93%) of the standard compound represent by Formula 5: melting point, 231- 233°C (decomposed); 1H-NMR (DMSOd6, 300 MHz), δ 1.9 (m, 4H), 2,7 (m, 2H), 4.0 (t, 2H), 7.0 (s, 2H), 7.1 (s, IH), 7.2-7.3 (m, 5H), 7.6 (t, IH), 7.9 (t, IH), 8.0 (m, 2H), 8.3 (t, IH), 10.0 (bs, IH).
Example 3: Synthesis of pranlukast
One hundred ml of methanol and 100 ml of methylene chloride (MC) were added to 10 g of N-(4-oxo-2-(l-trityl-lH-tetrazol-5-yl)-4H-chromen-8-yl)-4-(4- phenylbutoxy) benzamide (Pharmacostech), then 10 g of resin pre-treated with hydrochloric acid of pH 2-3 (TRILITE SCR-10 gel type) was added to the reaction mixture, followed by refluxing for 12 hours. The solid components were filtered out from the reaction mixture and washed with 100 ml of methanol. The filter-in solution was subject to vacuum distillation to obtain a solid substance and the solid was dissolved in 50 ml of dimethyl acetamide (DMAC). Afterwards, 200 ml of aqueous solution was added to the DMAC solution, and stirred for 1 hour at room temperature. Then the solid formed was filtered out, dried, and left for 5 hours at room temperature to obtain 6.18 g (yield rate: 93%) of the standard compound represent by Formula 5: melting point, 231-233°C (decomposed); 1H-NMR (DMSO-d6, 300 MHz), δ 1.9 (m, 4H), 2,7 (m, 2H), 4.0 (t, 2H), 7.0 (s, 2H), 7.1 (s, IH), 7.2-7.3 (m, 5H), 7.6 (t, IH), 7.9 (t, IH), 8.0 (m, 2H), 8.3 (t, IH), 10.0 (bs, IH).
Pranlukast and its hydrates come into the market as a capsule of Onon® Cap. (112.5 mg pranlukast hydrates/capsule, Dong-A Pharmaceutical).

The conventional method for preparing pranlukast was disclosed in US Pat. No. 5,587,483 and pranlukart is prepared by the following reaction formula I.
Reaction Formula I

As described in the reaction formula I, the acid chloride represented by formula 11 is obtained by reacting the benzoic derivative of formula 10 with the thionyl chloride. The resulting compound is reacted with the compound represented by formula 4. The compound (n = 4) represented by formula 5 is reacted with the tetrazol derivative represented by formula 6 to introduce tetrazol group and then benzopyran ring is formed, preparing pranlukast. However, the preparation method according to the reaction formula I has quite a few problems: (a) difficult manipulation due to utilizing excess amounts of toxic thionyl chlorides around a reflux temperature when the acid chloride represented by formula 11 is obtained by reacting the benzoic derivative of formula 10 with the thionyl chloride;
(b) hard elimination of thionyl chlorides toxic in a body after terminating the reactions; (c) requirement of base in an equivalent ratio of above 4 to collect the compound represented by formula 7; (d) unsuitability of massive production in a economical area because the compound is modified into a form of natrium salt and then purified for removal of contaminants after preparing pranlukart.
On the other hand, as described in the following reaction formula II in US Pat. No. 5,874,593, nitril compounds of formula 8 are reacted with hydrazine to prepare amidrazone compounds of formula 9a and 9b, and then pranlukart is fabricated by performing a tetrazol ring reaction using nitrous acids.
Reaction Formula II

However, the preparation method according to the reaction formula II has also the following difficulties: (a) it is difficult to perform the method due to utilizing excess amounts of toxic thionyl chlorides around a reflux temperature to obtain the acid chloride derivative in the preparation of the compounds represented by formula 8; (b) it is very difficult and toxic in body to eliminate thionyl chlorides after terminating the reactions; (c) it is not easy to massively produce the compounds of interest in an industrial-scale because much hydrazine toxic in body and nitrogen oxides harmful in environment are generated and unstable nitrous acids are used during the reactions.
Likewise, US Pat. No. 5,874,593, as described in the following reaction formula III, discloses that benzoic derivatives of formula 10′ are reacted with oxalyl chlorides to isolate acid chlorides represented by formula 11′, and the resulting acid chlorides are reacted with benzopyran amine derivatives containing tetrazol of formula 12, producing various derivatives containing pranlukart.
Reaction Formula III

( I D’ ] (H ‘ )

Oxalyl chlorides are massively used because the preparation method according to the reaction formula III is very expensive cost and has highly hygroscopic characteristics. In addition, the method has to be carried out under violent conditions that the temperature is increased up to around reflux temperature using 1,2- dichloroethanol as a solvent and further reacted for 1 hr. It is also difficult to remove harmful carbon monoxide and chlorine gases massively generated in elimination of oxalyl chloride after terminating the reactions, and it is not feasible to be applied into an industrial mass-production because the reaction is carried out under conditions of anhydrous and inactive gases
EXAMPLE 1: Preparation of Pranlukart Hemihydrates 4-(4-phenylbutoxy)benzoic acid (29.1 g; 1.1 equivalent ratio; prepared according to the method disclosed in US Pat. No. 4,780,469) was dissolved in 80 ml dimethylacetamide (DMAC, Aldrich) at 00C and then thionyl chloride (14.2 g, 1.2 equivalent ratio, Aldrich) was gradually added to the solution. After the mixture solution was stirred for 10 min at 00C, the mixture of 8-amino-4-oxo-tetrazol-5-yl-4H- 1-benzopyran hydrochloride salt (26.7 g; 1 equivalent ratio; prepared according to the method disclosed in US Pat. No. 4,780,469) and triethylamine (TEA, 10.1 g, 1 equivalent ratio, Aldrich) dissolved in 80 ml dimethylacetamide (DMAC, Aldrich) was slowly added to the mixture solution, and thermally stirred for 5 hrs at 25°C.
The reaction mixture was mixed with 300 ml H2O and stirred for 1 hr at 250C. The solid material obtained by filtering the solid material produced was washed with 100 ml H2O. 200 ml 50% acetone aqueous solution was added to the solid material and then refluxed for 1 hr. After the reaction mixture was cooled to room temperature, filtered and air-dried, the mixture was kept to stand on air for 5 hrs, obtaining 47.0 g pranlukart hemihydrates (yield rate: 98%): melting point 231-233°C (decomposition); 1H-NMR (DMSO-d6, 300 MHz) δ 1.9 (m, 4H), 2,7 (m, 2H), 4.0 (t, 2H), 7.0 (s, 2H), 7.1 (s, IH), 7.2-7.3 (m, 5H), 7.6 (t, IH), 7.9 (t, IH), 8.0 (m, 2H), 8.3 (t, IH), 10.0 (bs, IH).
EXAMPLE 2: Preparation of Pranlukart Hemihydrates – Substitution of the Chlorinating Agent
4-(4-phenylbutoxy)benzoic acid (29.1 g, 1.1 equivalent ratio) was dissolved in 80 ml dimethylacetamide (DMAC) at 00C and then oxalyl chloride (15.2 g, 1.2 equivalent ratio, Aldrich) was gradually added to the solution. After the mixture solution was stirred for 10 min at 00C, the mixture of 8-amino-4-oxo-tetrazol-5-yl-4H- 1-benzopyran hydrochloride salt (26.7 g; 1 equivalent ratio) and triethylamine (TEA, 10.1 g, 1 equivalent ratio) dissolved in 80 ml dimethylacetamide (DMAC) solution was slowly added to the mixture solution, and thermally stirred for 5 hrs at 25°C.
The reaction mixture was mixed with 300 ml H2O and stirred for 1 hr at 25°C. The solid material obtained by filtering the solid material produced was washed with 100 ml H2O. 200 ml 50% acetone aqueous solution was added to the solid material and then refluxed for 1 hr. After the reaction mixture was cooled to room temperature, filtered and air-dried, the mixture was kept to stand on air for 5 hrs, obtaining 43.3 g pranlukart hemihydrates (yield rate: 92%).
EXAMPLE 3: Preparation of Pranlukart Hemihydrates – Change of Base Condition
4-(4-phenylbutoxy)benzoic acid (29.1 g, 1.1 equivalent ratio) was dissolved in 80 ml dimethylacetamide (DMAC) at 00C and then thionyl chloride (14.2 g, 1.2 equivalent ratio) was gradually added to the solution. After the mixture solution was stirred for 10 min at 00C, the mixture of 8-amino-4-oxo-tetrazol-5-yl-4H-l-benzopyran hydrochloride salt (26.7 g; 1 equivalent ratio) and pyridine (7.9 g, 1 equivalent ratio, Aldrich) dissolved in 80 ml dimethylacetamide (DMAC) solution was slowly added to the mixture solution, and thermally stirred for 5 hrs at 25°C.
The reaction mixture was mixed with 300 ml H2O and stirred for 1 hr at 25°C. The solid material obtained by filtering the solid material produced was washed with 100 ml H2O. 200 ml 50% acetone aqueous solution was added to the solid material and then refluxed for 1 hr. After the reaction mixture was cooled to room temperature, filtered and air-dried, the mixture was kept to stand on air for 5 hrs, obtaining 45.6 g pranlukart hemihydrates (yield rate: 95%).
EXAMPLE 4: Preparation of Pranlukart Hemihydrates – Change of Reaction Temperature Condition
4-(4-phenylbutoxy)benzoic acid (29.1 g, 1.1 equivalent ratio) was dissolved in 80 ml dimethylacetamide (DMAC) at 00C and then thionyl chloride (14.2 g, 1.2 equivalent ratio) was gradually added to the solution. After the mixture solution was stirred for 10 min at O0C, the mixture of 8-amino-4-oxo-tetrazol-5-yl-4H-l-benzopyran hydrochloride salt (26.7 g; 1 equivalent ratio) and triethylamine (TEZ, 10.1 g, 1 equivalent ratio) dissolved in 80 ml dimethylacetamide (DMAC) solution was slowly added to the mixture solution, and thermally stirred for 4 hrs at 500C. The reaction mixture was mixed with 300 ml H2O and stirred for 1 hr at 500C. The solid material obtained by filtering the solid material produced was washed with 100 ml H2O. 200 ml 50% acetone aqueous solution was added to the solid material and then refluxed for 1 hr. After the reaction mixture was cooled to room temperature, filtered and air-dried, the mixture was kept to stand on air for 5 hrs, obtaining 45.6 g pranlukart hemihydrates (yield rate: 95%).
EXAMPLE 5: Preparation of Pranlukart Hemihydrates – Substitution of Reaction Solvent 4-(4-phenylbutoxy)benzoic acid (29.1 g, 1.1 equivalent ratio) was dissolved in 80 ml N-methylpyrrolidine (NMP, Aldrich) at O0C and then thionyl chloride (14.2 g, 1.2 equivalent ratio) was gradually added to the solution. After the mixture solution was stirred for 10 min at 00C, the mixture of 8-amino-4-oxo-tetrazol-5-yl-4H-l-benzopyran hydrochloride salt (26.7 g; 1 equivalent ratio) and triethylamine (TEZ, 10.1 g, 1 equivalent ratio) dissolved in 80 ml N-methylpyrrolidine (NMP) solution was slowly added to the mixture solution, and thermally stirred for 4 hrs at 250C. The reaction mixture was mixed with 300 ml H2O and stirred for 1 hr at 25°C.
The solid material obtained by filtering the solid material produced was washed with 100 ml H2O. 200 ml 50% acetone aqueous solution was added to the solid material and then refluxed for 1 hr. After the reaction mixture was cooled to room temperature, filtered and air-dried, the mixture was kept to stand on air for 5 hrs, obtaining 43.3 g pranlukart hemihydrates (yield rate: 90%).
EXAMPLE 6: Preparation of Pranlukart Hemihydrates – Equivalent Ratio Change of the Chlorinating Agent
4-(4-phenylbutoxy)benzoic acid (29.1 g, 1.1 equivalent ratio) was dissolved in 80 ml dimethylacetamide (DMAC) at 00C and then thionyl chloride (14.2 g, 1 equivalent ratio) was gradually added to the solution. After the mixture solution was stirred for 10 min at 00C, the mixture of 8-amino-4-oxo-tetrazol-5-yl-4H-l-benzopyran hydrochloride salt (26.7 g, 1 equivalent ratio) and triethylamine (TEZ, 10.1 g, 1 equivalent ratio) dissolved in 80 ml dimethylacetamide (DMAC) solution was slowly added to the mixture solution, and thermally stirred for 4 hrs at 25°C.
The reaction mixture was mixed with 300 ml H2O and stirred for 1 hr at 25°C. The solid material obtained by filtering the solid material produced was washed with 100 ml H2O. 200 ml 50% acetone aqueous solution was added to the solid material and then refluxed for 1 hr. After the reaction mixture was cooled to room temperature, filtered and air-dried, the mixture was kept to stand on air for 5 hrs, obtaining 44.6 g pranlukart hemihydrates (yield rate: 93%).
……………………..


 MONTELUKAST
MONTELUKAST| NPP | Mar 26, 2015 |
NPP=new patient population exclusivity

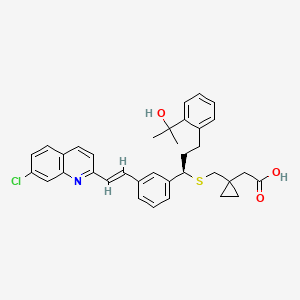













Montelukast sodium (I) is an active ingredient of products used for the treatment of respiration diseases, mainly asthma and nasal allergy. Montelukast sodium, chemically the sodium salt of [R-(E)]-l-[[[l-[3-[2-(7-chloro-2-quinolinyl)ethenyl]phenyl]-3-[2-(l-hydroxy-l- methylethyl)phenyl]propyl]thio]-methyl]cyclopropane acetic acid is described by the chemical formula (I).

(I)
The first solution of chemical synthesis of montelukast (I) was described in the patent no. EP 0480717 Bl and subsequently in specialized literature as well (M.Labele, Bioorg.Med.Chem.Lett. 5 (3), 283-288 (1995)). More possibilities of chemical synthesis of montelukast (I) are described in the following patents: EP 0480717 Bl, EP 0737186 Bl, US 2005/0234241 Al, WO 2005/105751 Al, US 2005/0107612 Al, WO 2005/105749 A2, WO 2005/105750 Al, US 2007/208178 Al.

(H) R alkyl

R-ι> R2 alkyl or hydrogen
For the process of isolation and purification of crude montelukast salts of montelukast with some amines (II) or montelukast acid (III) in the solid state have been used so far. Among montelukast salts with amines salts with dicyclohexylamine (EP 0737186 Bl, WO 04108679A1), tert-butylamine (US 2005/0107612 Al, WO 06043846A1), ethylphenylamine (US 2005/0107612 Al), isopropylamine (WO 2007/005965 Al), di-n-propylamine (WO 2007/005965 Al) and with cycloalkylamines (C5-C9, US 2007/213365 Al) have been described. Solid forms of montelukast acid, both crystalline and amorphous, have been described in a number of patent applications: WO 2005/040123, WO 2005/073194 A2, WO 2005/074893 Al, WO 2005/074893 Al, WO 2004/108679 Al, WO 2005/074935 Al. The most common method used in practice consists in purifying crude montelukast (I) via its salts with secondary amines, mainly with dicyclohexylamine (EP 0737186 Bl).
The sodium salt of montelukast, its preparation and various forms, amorphous or crystalline, , are described in a number of patents or patent applications, e.g. amorphous montelukast sodium is dealt with by EP 0737186 Bl, WO 03/066598 Al, WO 2004/108679 Al, WO 2005/074893 Al, WO 2006/054317A1 a WO 2007/005965. Crystalline polymorphs of montelukast sodium are described by WO 2004/091618 Al and WO 2005/075427 A2.
Processes of isolation and purification of montelukast are of crucial economic significance as they make it possible to obtain a substance that can be used for pharmaceutical purposes. These processes are used to remove impurities that result from the chemical instability of montelukast as well as the instability of the raw materials used for its chemical synthesis or non-selectivity of chemical reactions, or they may be represented by residues of the raw materials used, especially solvents. There is a general rule that chemical purity of the active pharmaceutical ingredient (API) produced in the industrial scale is one of the critical parameters for its commercialization. The American Food and Drug Administration (FDA) as well as European medicament control offices require, according to the Q7A ICH (International Conference on Harmonization) instruction, that API is freed from impurities to the maximum possible extent. The reason is achieving maximum safety of using the drug in the clinical practice. National inspection and control offices usually require that the content of an individual impurity in an API should not exceed the limit of 0.1%. All the substances (generally referred to as impurities) contained in an API over the limit of 0.1% should be isolated and characterized in accordance with the ICH recommendations. It is also recommended to isolate and characterize degradation products that are generated during the storage or usability period of API (ICH Guideline, 2006). In order to obtain information about the stability of a substance and to describe degradation products so-called “stress tests” are performed. Within these tests the API is subjected to a series of critical conditions the selection of which depends on the structure of the tested API. Usually, the influence of an increased temperature, air humidity, light, oxygen and stability in a wide pH range is assessed.
In the montelukast molecule there are a number of functional groups that impair the chemical stability of this substance. Montelukast is known to be prone to several types of degradation; it is mainly the case of three kinds of chemical transformation: (a) Oxidation of the mercapto group to the sulphoxide according to equation (1),

(b) Isomerisation at the location of the double bond from geometry (E) to (Z), or trans to cis by the effect of light according to equation (2),

(c) Dehydration at the location of tert. alcohol, producing the corresponding olefin according to equation (3).

Literature (E.D.Nelson, J.Pharm.Sci. 95, 1527-1539 (2006), C.Dufresne, J.Org.Chem. 1996, 61(24), 8518-8525, WO 2007005965A1) describes increased sensitivity of montelukast (or rather the mercapto group, which montelukast contains) to oxygen, see equation (I)). As the main product of oxidation of montelukast (I) (E)-montelukast-sulfoxide, chemically the sodium salt of [R-(E)]]-l-[[[l-[3-[2-(7-chloro-2-quinolinyl)ethenyl]phenyl]-3-[2-(l-hydroxy- l-methylethyl)-phenyl]propyl]sulfmyl]methyl]cyclopropane acetic acid, described with chemical formula (IV), is mentioned. Contamination of the product with this impurity is undesirable. For this reason the processes leading to the target substance are carried out with the exclusion of oxygen, i.e. under the protective atmosphere of an inert gas (e.g. nitrogen according to EP 0737186 Bl). (E)-Montelukast-sulfoxide (IV) has also been described as a product of the oxidative metabolism of montelukast (Balani S. K. et al: Drug Metabolism and Disposition (1997) 25 (11), 1282-87, Dufrense C: J.Org.Chem. (1996) 61(24), 8518-25).
Exposure of montelukast to light causes its isomerization while a montelukast derivative with geometry (Z) is generated in the location of the double bond (Smith Glen A. et al: Pharm.Res. 2004, 21(9), 1539-44). The impurity resulting from photo-instability is (Z)-montelukast, chemically the sodium salt of l-[[[(lR)-l-[3-[(lZ)-2-(7-chloro-2-quinolinyl)ethenyl]phenyl]- 3-[2-(l-hydroxy-l-methylethyl)phenyl]propyl]thio]methyl]cyclopropane acetic acid, which is described by chemical formula (V), see equation (2).
Another degradation impurity described in literature (WO 2007005965A1) is montelukast dehydrated, chemically the sodium salt of l-[[[(lR)-l-[3-[(lE)-2-(7-chloro-2- quinolinyl)ethenyl]-phenyl]-3-[2-(l-methylethenyl)phenyl]propyl]thio]methyl]cyclopropane acetic acid, described by chemical formula (VI), see equation (3).

Recently, montelukast or its pharmaceutically acceptable salt is known to function as an antagonist and also as a biosynthesis inhibitor against leukotrienes. The sodium salt of montelukast is commercially available from Merck under the trademark of Singulair® for treating asthma.
EP 480,717 discloses a method of preparing said montelukast sodium salt: As shown in Reaction Scheme 1, methyl 1-(mercaptomethyl)cyclopropylacetate of formula (B) is coupled with the compound of formula (A) to produce the compound of formula (C) as an intermediate, and the compound of formula (C) is then hydrolyzed to obtain the free acid form thereof, followed by treating the free acid with NaOH. However, this method gives a low yield or the manufacturing cost is high.

THP: tetrahydropyranyl
PPTS: Pyridinium p-toluenesulfonateIn order to solve the above-mentioned problems, EP 737,186 suggests a method as shown in Reaction Scheme 2. This method uses a methanesulfonyl compound of formula (A′) having an unprotected hydroxyl group instead of the THP-protected compound of formula (A). Further, this method uses 1-(mercaptomethyl)cyclopropylacetate dilithium salt of formula (B′) instead of methyl 1-(mercaptoethyl)cyclopropylacetate of formula (B), thereby making the subsequent deprotection step unnecessary. Subsequently, dicyclohexylamine is added to the compound of formula (C″) to produce the compound of formula (D), which is converted to the desired sodium salt.


However, the methanesulfonyl compound of formula (A′) used in the above process as a starting material is very unstable, which makes the whole process very complicated. Namely, the reaction to produce the compound of formula (A′) must be performed at a low temperature of about −30° C. and the product is required to be kept at about −15° C. The compound of formula (A′) thus produced is unstable toward moisture and air, and therefore, the reaction thereof has to be conducted quickly under carefully controlled conditions. Also, the synthesis of the compound of formula (B′) requires the use of n-butyllithium which is very explosive and unstable toward moisture and air. Thus, the method described in Reaction Scheme is not suitable for large-scale production.
Example 2Preparation of 1-(((1-(R)-(3-(2-(7-chloro-2-quinolidyl)-ethenyl)phenyl)-3-(2-(1-hydroxy-1-methyl-ethyl)phenyl)propyl)thio)methyl)-cyclopropylacetic acid12.7 g of 1-(mercaptomethyl)cyclopropylacetic acid dissolved in 90 ml of dimethylformamide was slowly added to a solution of 6.26 g of 60% sodium hydride dissolved in 90 ml of dimethylformamide at 0 to 5° C. To the resulting mixture, 30 g of 2-(2-(3-(S)-(3-(2-(7-chloro-2-quinolinyl)ethenyl)phenyl)-3-diphenylphosphate oxypropyl)phenyl)-2-propanol obtained in Example 1 dissolved in 120 ml of dimethylformamide was slowly added dropwise. After the temperature was slowly increased to room temperature, the reaction was run for 18 to 20 hrs. Then, the reaction mixture was neutralized with a saturated ammonium chloride aqueous solution, and treated with ethyl acetate and distilled water. With shaking, the organic layer was separated and dried over sodium sulfate, followed by removing the solvent under reduced pressure. The residue thus obtained was dissolved in 270 ml of cyclohexane, and the product was recrystallized therefrom. The crystallized product was filtered, washed and dried to obtain 22.2 g (87.1%) of the title compound as a yellow solid.
1H-NMR (300 MHz, CD3OD): δ 8.27 (1H, d), 7.98 (1H, s), 7.78 (2H, d), 7.73 (2H, d), 7.38-7.56 (6H, m), 7.07-7.14 (3H, m), 4.84 (1H, t), 3.30-3.33 (1H, m), 2.84-2.87 (1H, m), 2.52 (2H, s), 2.41 (2H, s), 2.18-2.23 (2H, m), 1.55 (6H, s), 0.37-0.52 (4H, m).
Example 3Preparation of 1-(((1-(R)-(3-(2-(7-chloro-2-quinolidyl)ethenyl)phenyl)-3-(2-(1-hydroxy-1-methyl-ethyl)phenyl)propyl)-thio)methyl)cyclopropylacetate sodium saltStep 1: Preparation of methyl 1-(((1-(R)-(3-(2-(7-chloro-2-quinolidyl)-ethenyl)phenyl)-3-(2-(1-hydroxy-1-methyl-ethyl)phenyl)propyl)thio)methyl)-cyclopropylacetate
2.1 g of methyl 1-(acetylthiomethyl)cyclopropylacetate dissolved in 35 ml of dimethylformamide was slowly added to a solution of 0.71 g of 60% sodium hydride dissolved in 35 ml of dimethylformamide at a temperature ranging from 0 to 5° C. To the resulting mixture, 7.73 g of 2-(2-(3-(S)-(3-(2-(7-chloro-2-quinolinyl)ethenyl)phenyl)-3-diphenylphosphate oxypropyl)phenyl)-2-propanol obtained in Example 1 dissolved in 35 ml of dimethylformamide was slowly added dropwise at a temperature ranging from 0 to 5° C. After about 1 hr, the reaction mixture was treated with ethyl acetate and distilled water. With shaking, the organic layer was separated and dried over sodium sulfate, followed by removing the solvent under reduced pressure to obtain 5.68 g (84.5%) of the title compound as a yellow liquid.
1H-NMR (300 MHz, CDCl3): δ 8.12 (2H, d), 7.66-7.74 (4H, m), 7.37-7.48 (6H, m), 7.12-7.20 (3H, m), 3.96 (1H, t), 3.14-3.16 (1H, m), 2.88 (1H, m), 2.53 (2H, s), 2.43 (2H, s), 1.62 (6H, d), 0.41-0.54 (4H, m).
Step 2: Preparation of 1-(((1-(R)-(3-(2-(7-chloro-2-quinolidyl)-ethenyl)phenyl)-3-(2-(1-hydroxy-1-methyl-ethyl)phenyl)propyl)thio)methyl)-cyclopropyl acetic acid
12 g of methyl 1-(((1-(R)-(3-(2-(7-chloro-2-quinolidyl)ethenyl)phenyl)-3-(2-(1-hydroxy-1-methyl-ethyl)phenyl)propyl)thio)methyl)cyclopropylacetate obtained in step 1 was dissolved in a mixture of 60 ml of tetrahydrofuran and 30 ml of methyl alcohol. After adjusting the temperature to 10 to 15° C., 24 g of 10% NaOH solution was slowly added to the resulting mixture. Then, the temperature was slowly increased to room temperature (24 to 27° C.), and the reaction mixture was stirred for 20 hrs. After reaction was completed, the organic layer was separated and dried, followed by removing the solvent under reduced pressure. The residue thus obtained was mixed with water layer again, and 120 ml of toluene was added thereto. Subsequently, the pH of the reaction product was adjusted to 4 by adding 300 ml of acetic acid. The organic layer was separated again and dried over sodium sulfate, followed by removing the solvent under reduced pressure. The residue thus obtained was dissolved in 96 ml of a mixture of isopropanol and distilled water (2:1), and the product was recrystallized therefrom. The crystallized product was filtered to obtain 9.82 g (83%) of the title compound as a yellow solid.
Montelukast acid
1H-NMR (300 MHz, CD3OD): δ 8.27 (1H, d), 7.98 (1H, s), 7.78 (2H, d), 7.73 (2H, d), 7.38-7.56 (6H, m), 7.07-7.14 (3H, m), 4.84 (1H, t), 3.30-3.33 (1H, m), 2.84-2.87 (1H, m), 2.52 (2H, s), 2.41 (2H, s), 2.18-2.23 (2H, m), 1.55 (6H, s), 0.37-0.52 (4H, m).
m.p.: 154° C., purity>99%
Step 3: Preparation of 1-(((1-(R)-(3-(2-(7-chloro-2-quinolidyl)ethenyl)phenyl)-3-(2-(1-hydroxy-1-methyl-ethyl)phenyl)propyl)thio)-methyl)cyclopropylacetate sodium salt
5 g of 1-(((1-(R)-(3-(2-(7-chloro-2-quinolidyl)ethenyl)phenyl)-3-(2-(1-hydroxy-1-methyl-ethyl)phenyl)propyl)thio)methyl)cyclopropylacetic acid obtained in step 2 was mixed with 10 ml of toluene, followed by removing the solvent under reduced pressure to remove the solvent. To the residue thus obtained, 14.5 ml of toluene and 13 ml of 0.5N NaOH/MeOH solution were sequentially added. The resulting mixture was stirred for 30 min, followed by removing the solvent under reduced pressure. The residue was dissolved in 10 ml of toluene and 50 ml of n-hexane, and the product was recrystallized therefrom. The crystallized product was filtered to obtain 5.1 g (98%) of the title compound as a pale yellow solid.
Montelukast sodium
1H-NMR (300 MHz, CD3OD): δ 8.29 (1H, d), 7.99 (1H, s), 7.83-7.91 (3H, m), 7.72 (1H, s), 7.49-7.52 (2H, m), 7.38-7.44 (4H, m), 7.10-7.15 (3H, m), 4.04 (1H, t), 3.08 (1H, m), 2.82 (1H, m), 2.66 (1H, d), 2.52 (1H, d), 2.43 (1H, d), 2.29 (1H, d), 2.16-2.24 (2H, m), 1.52 (6H, s), 0.33-0.52 (4H, m)
| WO1995018107A1 | Dec 22, 1994 | Jul 6, 1995 | James J Bergan | Process for the preparation of leukotriene antagonists |
| WO2004026838A1 * | Sep 11, 2003 | Apr 1, 2004 | Michiaki Adachi | Method for producing a 3,5-dihydroxy-6-heptenoate |
| WO2009111998A2 * | Mar 11, 2009 | Sep 17, 2009 | Zentiva, K.S. | Specific impurities of montelukast |
| EP0480717A1 | Oct 10, 1991 | Apr 15, 1992 | Merck Frosst Canada Inc. | Unsaturated hydroxyalkylquinoline acids as leukotriene antagonists |
| EP0480717B1 | Oct 10, 1991 | Apr 15, 1998 | Merck Frosst Canada Inc. | Unsaturated hydroxyalkylquinoline acids as leukotriene antagonists |
| EP0737186B1 | Dec 22, 1994 | Aug 19, 1998 | Merck & Co., Inc. | Process for the preparation of leukotriene antagonists |
| US2985589 | May 22, 1957 | May 23, 1961 | Universal Oil Prod Co | Continuous sorption process employing fixed bed of sorbent and moving inlets and outlets |
| US5156736 | May 7, 1991 | Oct 20, 1992 | Schoenrock Karlheinz W R | Simulated moving bed apparatus using a single sorbent bed for separating components from a fluid stream |
| US5523477 | Jan 23, 1995 | Jun 4, 1996 | Merck & Co., Inc. | Reacting 1,1-cyclopropanedimethanol with dialkyl sulfite in presence of acid or base to form cyclic sulfite, removing alcohol reaction by-product |
| US5565473 | Feb 23, 1995 | Oct 15, 1996 | Merck Frosst Canada, Inc. | Useful as anti-asthmatic, anti-allergic, anti-inflammatory and cytoprotective agents; montelukast and its sodium salt |
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2006008751A2 * | Jul 19, 2004 | Jan 26, 2006 | Satyanarayana Chava | Process for the preparation of montelukast and its salts |
| WO2006043846A1 * | Oct 21, 2005 | Apr 27, 2006 | Inst Farmaceutyczny | Salt of montelukast with tert.-butylamine |
| WO2007072114A1 * | Jan 16, 2006 | Jun 28, 2007 | Harmander Pal Singh Chawla | An improved process for the manufacture of montelukast sodium |
| WO2007107297A1 * | Mar 15, 2007 | Sep 27, 2007 | Synthon Bv | Montelukast amantadine salt |
| US20050107612 * | Dec 30, 2003 | May 19, 2005 | Dr. Reddy’s Laboratories Limited | Process for preparation of montelukast and its salts |
| Reference | ||
|---|---|---|
| 1 | * | “An improved process to obtain Montelukast sodium” RESEARCH DISCLOSURE, MASON PUBLICATIONS, HAMPSHIRE, GB, vol. 521, no. 2, 1 September 2007 (2007-09-01), page 908, XP007137576 ISSN: 0374-4353 |
| 2 | * | “Piperazine salts of Montelukast, a new efficient method of purification” IP.COM JOURNAL, IP.COM INC., WEST HENRIETTA, NY, US, 29 November 2007 (2007-11-29), XP013122974 ISSN: 1533-0001 |
| 3 | * | AL OMARI ET AL: “Effect of light and heat on the stability of montelukast in solution and in its solid state” JOURNAL OF PHARMACEUTICAL AND BIOMEDICAL ANALYSIS, NEW YORK, NY, US, vol. 45, no. 3, 19 October 2007 (2007-10-19), pages 465-471, XP022306740 ISSN: 0731-7085 |
| 4 | * | DUFRESNE C ET AL: “Synthesis of montelukast (MK-0476) metabolic oxidation products” JOURNAL OF ORGANIC CHEMISTRY, AMERICAN CHEMICAL SOCIETY, EASTON.; US, vol. 61, no. 24, 1 January 1996 (1996-01-01), pages 8518-8525, XP002284162 ISSN: 0022-3263 |
| 5 | * | GRAUL L ET AL: “Montelukast sodium, MK-476, MK-0476, L-706631, Singulair” DRUGS OF THE FUTURE, PROUS SCIENCE, ES, vol. 22, no. 10, 1 January 1997 (1997-01-01), page 1103, XP008082254 ISSN: 0377-8282 |
| 6 | * | NELSON ERIC D ET AL: “Evaluation of solution oxygenation requirements for azonitrile-based oxidative forced degradation studies of pharmaceutical compounds.” July 2006 (2006-07), JOURNAL OF PHARMACEUTICAL SCIENCES JUL 2006, VOL. 95, NR. 7, PAGE(S) 1527 – 1539 , XP002563008 ISSN: 0022-3549 compound 4 |
| 7 | * | SMITH GLENN A ET AL: “An automated method for the determination of montelukast in human plasma using dual-column HPLC analysis and peak height summation of the parent compound and its photodegradation product.” September 2004 (2004-09), PHARMACEUTICAL RESEARCH SEP 2004, VOL. 21, NR. 9, PAGE(S) 1539 – 1544 , XP002563007 ISSN: 0724-8741 page 1539 – page 1544; example 2 |
| WO2011061545A1 * | Nov 20, 2010 | May 26, 2011 | Generics [Uk] Limited | Hplc method for analyzing vorinostat |
| WO2012077123A1 * | May 12, 2011 | Jun 14, 2012 | Arch Pharmalabs Limited | Purification of montelukast using a simulated moving bed |
| WO2014034203A1 * | May 28, 2013 | Mar 6, 2014 | Dai Nippon Printing Co., Ltd. | Method for producing high-purity montelukast |
| CN102060762A * | Jan 28, 2011 | May 18, 2011 | 海南美大制药有限公司 | Montelukast compound and new preparation method thereof |
| CN102060762B | Jan 28, 2011 | May 29, 2013 | 海南美大制药有限公司 | Montelukast compound and new preparation method thereof |
| US8471030 | Dec 6, 2010 | Jun 25, 2013 | Orochem Technologies Inc. | Purification of montelukast using simulated moving bed |
| US8754129 | Nov 25, 2009 | Jun 17, 2014 | Generics [Uk] Limited | Crystalline vorinostat form VI |

USV Limited
16-Oct-2014 pub date
WO-2014167577-A2
 PATENT FEATURE ON THIS BLOG
PATENT FEATURE ON THIS BLOG
Title of the invention: “”SYNTHESIS OF DABIGATRAN”.”
Applicants: USV LIMITED (IN).
Inventors: Laxmikant Narhari Patkar (IN), Harish Kashinath Mondkar (IN), Sachin Shivaji Patil (IN), Tanaji Shamrao Jadhav (IN), Nitin Nivrutti Hagavane (IN), Rajesh Ganpat Bopalkar (IN) and Nitin Dnyaneshwar Arote (MY).

The present invention relates to a process for preparation of Dabigatran etexilate or pharmaceutically acceptable salt thereof. The present invention relates to novel compounds, in particular Ethyl-3-{[(2-formyl-l-methyl-lH-benzimidazole-5-yl) carbonyl] -(2-pyridinyl) amino} propanoate and Ethyl-3-{[(2-dichloromethyl-l-methyl -lH-benzimidazole-5-yl)carbonyl]- (2-pyridinyl) amino}propanoate and process for preparation thereof. The present invention further relates to the use of these novel compounds in the preparation of Dabigatran etexilate or pharmaceutically acceptable salt thereof.
Dabigatran is used to prevent strokes in those with atrial fibrillation due to non heart valve causes



![]()
Process for preparing dabigatran etexilate mesylate, useful for treating thrombosis, stroke and embolism. Also claims novel intermediates of dabigatran and their synthesis. Represents the first patenting from USV on this API, which was originally developed and launched, by Boehringer Ingelheim for treating conditions such as stroke, thrombosis and atrial fibrillation.
Dabigatran (Pradaxa in Australia, Europe and USA, Pradax in Canada, Prazaxa in Japan) is an oral anticoagulant from the class of the direct thrombin inhibitors. It is being studied for various clinical indications and in some cases it offers an alternative to warfarin as the preferred orally administered anticoagulant (“blood thinner”) since it does not require frequent blood tests for international normalized ratio (INR) monitoring while offering similar results in terms of efficacy. There is no specific way to reverse the anticoagulant effect of dabigatran in the event of a major bleeding event, unlike warfarin, although a potential dabigatran antidote (pINN: idarucizumab) is undergoing clinical studies. It was developed by the pharmaceutical company Boehringer Ingelheim.

Family members of its product case, WO9837075, have SPC protection in most EU states until February 2023, and expiry dates in the US until July 2020. The FDA Orange Book lists US7932273 (product derivative) and US7866474 (describing film blister-card containers for pradaxa®), which expire in September 2025 and August 2027 respectively, for dabigatran.
The drug also has New Chemical Entity exclusivity expiring on October 19, 2015. As of October 2014, Newport Premium™ reports that USV has dabigatran under development.
SEE
After looking through a number of flow articles that describe and illustrate processes toward the production of drug final products and advanced intermediates, I thought an article from Florida State — Tyler McQuade (open source Beilstein JOC 2013) was informative and storytelling. He was able to show some of the challenges that go into designing a flow methodology around process that have already been worked out in batch mode, and had been looked at in a number of labs already.
Before talking about the chemistry, Professor McQuade talks about a number of concerns in transferring technology from batch to flow: DOE, solvent exchange (precipitation and moving from one reaction to another), Cost of Goods Analysis – reaction concentrations, solvent costs, process time, by-product formation and purification. There certainly is a lot that goes into the strategy. To give you the framework: this group was looking to make a continuous process…
View original post 247 more words
DRUG REGULATORY AFFAIRS INTERNATIONAL

System Suitability for USP Chromatographic Methods
How should system suitability tests (SSTs) be structured for USP monographs? More about USP experts group’s recommendations on the parameters and acceptance criteria for SSTs and the essential aspects of this new approach can be found in this News.
read
An interesting article from the USP experts group “Small Molecules” has been published in the Pharmacopoeial Forum 39(5). It deals with USP’s future requirements regarding system suitability tests (SST).
SSTs are performed each time an analytical method is used. Together with instruments qualification and methods validation, the SST ensures the quality of analytical test results. The SST shows that a procedure and an instrumental system are performing as they did when the procedure was validated and that the method is thus “fit for purpose” for the intended use.
General requirements can be found in the USP Chapter <621> Chromatography which also contains provisions and acceptance…
View original post 116 more words
 Nintedanib
Nintedanib
FDA approves Ofev to treat idiopathic pulmonary fibrosis
10/15/2014
The U.S. Food and Drug Administration today approved Ofev (nintedanib) for the treatment of idiopathic pulmonary fibrosis (IPF).
read at
see synthesis
https://newdrugapprovals.org/2014/05/21/in-battle-of-ipf-drugs-bis-nintedanib-impresses/


The U.S. Food and Drug Administration today approved Esbriet (pirfenidone)
for the treatment of idiopathic pulmonary fibrosis (IPF).
read at
Click for synthesis



//////////
![]()
US priority review for Eisai cancer drug lenvatinib
Eisai has been boosted by news that regulators in the USA have agreed to a quicker review of its anticancer agent lenvatinib.
The US Food and Drug Administration has granted a priority review to Eisai’s New Drug Application for lenvatinib as a treatment for progressive radioiodine-refractory differentiated thyroid cancer. This means that the agency has assigned a Prescription Drug User Fee Act action date of April 14 next year, eight months after the NDA was submitted.
Read more at: http://www.pharmatimes.com/Article/14-10-15/US_priority_review_for_Eisai_cancer_drug_lenvatinib.aspx#ixzz3GH3iXiDU
SEE SYNTHESIS




NEW DRUG APPROVALS BLOG ON A HIGH
ALL ABOUT DRUGS, LIVE, BY DR ANTHONY MELVIN CRASTO, WORLDDRUGTRACKER, HELPING MILLIONS, 7 MILLION HITS ON GOOGLE, PUSHING BOUNDARIES, ONE LAKH PLUS CONNECTIONS WORLDWIDE, 4 LAKHS PLUS VIEWS ON THIS BLOG IN 198 COUNTRIES

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D

