
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
BENDAMUSTINE


TREANDA contains bendamustine hydrochloride, an alkylating drug, as the active ingredient. The chemical name of bendamustine hydrochloride is 1H-benzimidazole-2-butanoic acid, 5-[bis(2-chloroethyl)amino]-1 methyl-, monohydrochloride. Its empirical molecular formula is C16H21Cl2N3O2 • HCl, and the molecular weight is 394.7. Bendamustine hydrochloride contains a mechlorethamine group and a benzimidazole heterocyclic ring with a butyric acid substituent, and has the following structural formula:
|
TREANDA (bendamustine hydrochloride) for Injection is intended for intravenous infusion only after reconstitution with Sterile Water for Injection, USP, and after further dilution with either 0.9% Sodium Chloride Injection, USP, or 2.5% Dextrose/0.45% Sodium Chloride Injection, USP. It is supplied as a sterile non-pyrogenic white to off-white lyophilized powder in a single-use vial. Each 25-mg vial contains 25 mg of bendamustine hydrochloride and 42.5 mg of mannitol, USP. Each 100-mg vial contains 100 mg of bendamustine hydrochloride and 170 mg of mannitol, USP. The pH of the reconstituted solution is 2.5 -3.5.
Bendamustine hydrochloride, 4-{5-[Bis(2-chloroethyl) amino]- l-methyl-2- benzimidazolyl} butyric acid hydrochloride, of the formula (VI) :

was initially synthesized in 1963 in the German Democratic Republic (GDR) and was available from 1971 to 1992 there, as the hydrochloride salt, under the trade name Cytostasan®. Since that time, it has been marketed in Germany under the trade name Ribomustin®. Bendamustine Hydrochloride as injection is available in the United States under the tradename Treanda®. Bendamustine hydrochloride is an alkylating agent that is approved for the treatment of non-Hodgkin’s lymphoma, multiple myeloma and chronic lymphocytic leukemia.
Bendamustine hydrochloride is a benzimidazole analog. While bendamustine has been demonstrated as efficacious, it is known to be unstable, especially in aqueous solutions, leading to formation of non-bendamustine products (i.e. “degradation impurities”) which leads to technical difficulties in its preparation and administration. In light of its instability in aqueous solution, bendamustine is supplied as a lyophilized cake of bendamustine hydrochloride salt. US2006/159713, US 2006/128777 and WO2010/036702 disclose various impurities of Bendamustine hydrochloride which are as follows:

PC-1 PC-2
Jena et al. were the first to disclose the synthesis of Bendamustine hydrochloride in German (GDR) Patent No. 34727. Krueger et al. in German (GDR) Patent No. 159877 recite a method as summarized in scheme-1, for the synthesis of bendamustine hydrochloride comprising the reaction of the 4-[l-methyl-5-bis-(2- hydroxyethyl)-benzimidazolyl-2]butyric acid ethyl ester (4) (or the corresponding methyl, propyl or butyl ester) with thionyl chloride in chloroform at 0-5°C to form 4-[l- methyl-5-bis-(2-chloroethyl)-benzimidazolyl-2]butyric acid ethyl ester (5). Excess of thionyl chloride is destroyed by stirring the reaction mixture in aqueous HCl. Finally chloroform is distilled off and stirred at 95°C for 3 hours. The reaction mixture is partially concentrated and the residue is diluted with water and stirred upto crystallization. Further purification is done by recrystallization from water.
Scheme-1: Method disclosed by Krueger et al. in DD159877 for the synthesis of Bendamustine hydrochloride

Bendamustine hydrochloride (6)
Ozegowski et al in Zentralblatt fuer Pharmazie, Pharmakotherapie und Laboratoriumsdiagnostik 1 10 (10), 1013-1019 (1971) discloses a process for the preparation of bendamustine hydrochloride monohydrate. The Chinese journal “Chinese journal of New Drugs “, 2007, No. 23, Vol. 16, 1960-61 and J. Prakt. Chem. 20, 178-186 (1963) disclose another method for the synthesis of Bendamustine hydrochloride monohydrate starting from 2,4-dinitrochlorobenzene as summarized in scheme-2.

The crucial conversions are reaction of l-methyl-2-(4′-ethyl butyrate)-5- amino]-lH-benzimidazole 6 with ethylene oxide in the presence of water, sodium acetate and acetic acid, by maintaining at 5°C for 5 hours and overnight at 20°C to give 4-{5-[bis-(2-hydroxy-ethyl)-amino]-l-methyl-lH-benzimidazol-2-yl}-butyric acid ethyl ester (dihydroxy ester) 7 as a jelly mass, which on chlorination using thionyl chloride in chloroform and subsequent in situ hydrolysis with concentrated HCI gave bendamustine hydrochloride. It also discloses a process for the recrystallization of bendamustine hydrochloride from water and the product obtained is a monohydrate with a melting point of 148-151°C.
IP.com Journal 2009, 9(7B), 21 discloses another process as shown below for the preparation of ethyl-4-[5-[bis(2-hydroxyethyl) amino]- l-methylbenzimidazol-2- yl]butanoate (III) wherein ethyl-4-(5 -amino- 1 -methyl- lH-benzo[d]imidazol-2-yl) butanoate (II) is reacted with 2-halo ethanol in the presence of an inorganic base selected from the group consisting potassium carbonate, potassium bicarbonate, sodium

The PCT application WO 2010/042568 assigned to Cephalon discloses the synthesis of Bendamustine hydrochloride as summarized in schem-3 starting from 2,4- dintroaniline in six steps. The crucial step is reductive alkylation of Il-a, using borane- tetrahydrofuran and chloroacetic acid at ambient temperature, producing compound of formula I-a. Acid mediated hydrolysis of I-a using concentrated hydrochloric acid at reflux produced bendamustine hydrochloride which has a purity of 99.1%. The above PCT Patent application also discloses a method of purification of Bendamustine hydrochloride by agitating the Bendamustine hydrochloride in a mixture of DMF and THF at 75°C for about 30 minutes followed by cooling to ambient temperature and isolating the solid by filtration.
Scheme-3:


iil-a


Bemdamuatine hydrochloride
The PCT application WO 2011/079193 assigned to Dr. Reddy’s Laboratories discloses the synthesis of Bendamustine hydrochloride as summarized in schem-4 starting from compound of formula (II). The crucial step is alkylation of compound of formula II with 2-haloethanol in the presence of an organic base to give a compound of formula (III) which on chlorination with a chlorinating agent affords a compound of formula (IV). Compound of formula (IV) on hydrolysis in acidic medium gives bendamustine hydrochloride. It further discloses purification of bendamustine hydrochloride using aqueous hydrochloric acid and acetonitrile.
Scheme-4:

Bendamustine hydrochloride (Pure)
The most of the prior art processes described above involve
• The use of ethylene oxide for the preparation of bendamustine hydrochloride, which is often not suitable for industrial scale processes due to difficulty in handling ethylene oxide, since it is shipped as a refrigerated liquid.
• Further, the known processes involve the use of strongly acidic conditions and high temperatures for the hydrolysis of ethyl ester of bendamustine and subsequent in-situ formation of bendamustine hydrochloride, thereby resulting in increased levels of various process-related impurities IMP. -A (RRT-0.46), IMP. -B (RRT-1.27) and IMP. -C (RRT-1.31) whose removal is quite difficult and make the process less economically viable.

IMP.-B
International Application Publication No. WO 2009/120386 describes various solid forms of bendamustine hydrochloride designated as bendamustine hydrochloride Form 1, bendamustine hydrochloride Form 2, bendamustine hydrochloride Form 3, bendamustine hydrochloride Form 4, amorphous bendamustine hydrochloride or a mixture thereof, processes for their preparation and lyophilized composition comprising the solid forms. According to the disclosure, monohydrate of bendamustine hydrochloride has been prepared previously. The monohydrate has a reported melting point of 152-156°C which is similar to that of the observed melting point of bendamustine hydrochloride Form 2.
It is known that synthetic compounds can contain extraneous compounds or impurities resulting from their synthesis or degradation. The impurities can be unreacted starting materials, by-products of the reaction, products of side reactions, or degradation products. Generally, impurities in an active pharmaceutical ingredient (API) may arise from degradation of the API itself, or during the preparation of the API. Impurities in Bendamustine hydrochloride or any active pharmaceutical ingredient (API) are undesirable and might be harmful.
Regulatory authorities worldwide require that drug manufacturers isolate, identify and characterize the impurities in their products. Furthermore, it is required to control the levels of these impurities in the final drug compound obtained by the manufacturing process and to ensure that the impurity is present in the lowest possible levels, even if structural determination is not possible. The product mixture of a chemical reaction is rarely a single compound with sufficient purity to comply with pharmaceutical standards. Side products and byproducts of the reaction and adjunct reagents used in the reaction will, in most cases, also be present in the product mixture. At certain stages during processing of the active pharmaceutical ingredient, the product is analyzed for purity, typically, by HPLC, TLC. or GC analysis, to determine if it is suitable for continued processing and, ultimately, for use in a pharmaceutical product. Purity standards are set with the intention of ensuring that an API is as free of impurities as possible, and, thus, are as safe as possible for clinical use. The United States Food and Drug Administration guidelines recommend that the amounts of some impurities are limited to less than 0.1 percent.
Generally, impurities are identified spectroscopically and by other physical methods, and then the impurities are associated with a peak position in a chromatogram (or a spot on a TLC plate). Thereafter, the impurity can be identified by its position in the chromatogram, which is conventionally measured in minutes between injection of the sample on the column and elution of the particular component through the detector, known as the “retention time” (“RT”). This time period varies daily based upon the condition of the instrumentation and many other factors. To mitigate the effect that such variations have upon accurate identification of an impurity, practitioners use “relative retention time” (“RRT”) to identify impurities. The RRT of an impurity is its retention time divided by the retention time of a reference marker.
It is known by those skilled in the art, the management of process impurities is greatly enhanced by understanding their chemical structures and synthetic pathways, and by identifying the parameters that influence the amount of impurities in the final product.
Therefore, there remains a need for improved process for the preparation of bendamustine hydrochloride, producing high yield and purity, and well-suited for use on an industrial scale. Despite the existence of various polymorphic forms of bendamustine hydrochloride, there exists a need for a simple process for the preparation of the stable form of bendamustine hydrochloride which is amenable to scale up and results in high yield and purity.
Bendamustine (INN, trade names Treakisym, Ribomustin, Levact and Treanda; also known as SDX-105) is a nitrogen mustardused in the treatment of chronic lymphocytic leukemia[1] and lymphomas. It belongs to the family of drugs called alkylating agents. It is also being studied for the treatment of sarcoma.[2] It is also being investigated in phase II trials for the non-cancer treatment of AL Amyloidosis.
Bendamustine hydrochloride, initially synthesized in 1963 in the German Democratic Republic, is an alkylating agent that has been shown to have therapeutic utility in treating diseases such as chronic lymphocytic leukemia, Hodgkin’s disease, non-Hodgkin’s lymphoma, multiple myeloma, and breast cancer.It was available from 1971 to 1992 under the trade name Cytostasanand, since that time, has been marketed in Germany as Ribomustin.In March 2008 the FDA approved bendamustine hydrochloride under the trade name Treanda for the treatment of chronic lymphocytic leukemia (CLL). Approval for use in indolent B-cell non-Hodgkin’s lymphoma (NHL) was received in 2009.
History
Bendamustine was first synthesized in 1963 by Ozegowski and Krebs in East Germany (the former German Democratic Republic). Until 1990 it was available only in East Germany. East German investigators found that it was useful for treating chronic lymphocytic leukemia, Hodgkin’s disease, non-Hodgkin’s lymphoma, multiple myeloma and lung cancer.
Bendamustine received its first marketing approval in Germany, where it is marketed under the tradename Ribomustin, by Astellas Pharma GmbH’s licensee, Mundipharma International Corporation Limited. It is indicated as a single-agent or in combination with other anti-cancer agents for indolent non-Hodgkin’s lymphoma, multiple myeloma, and chronic lymphocytic leukemia. SymBio Pharmaceuticals Ltd holds exclusive rights to develop and market bendamustine HCl in Japan and selected Asia Pacific Rim countries.
In March 2008, Cephalon received approval from the United States Food and Drug Administration to market bendamustine in the US, where it is sold under the tradename Treanda, for treatment of chronic lymphocytic leukemia.[3]
In October 2008, the FDA granted further approval to market Treanda for the treatment of indolent B-cell non-Hodgkin’s lymphoma that has progressed during or within six months of treatment with rituximab or a rituximab-containing regimen.[4]
Pharmacology
Bendamustine is a white, water soluble microcrystalline powder with amphoteric properties. It acts as an alkylating agent causing intra-strand and inter-strand cross-links between DNA bases.
After intravenous infusion it is extensively metabolised in the liver by cytochrome p450. More than 95% of the drug is bound to protein – primarily albumin. Only free bendamustine is active. Elimination is biphasic with a half-life of 6–10 minutes and a terminal half-life of approximately 30 minutes. It is eliminated primarily through the kidneys. This paragraph is inconsistent with sidebar for primary excretion pathway.
Chemotherapeutic uses
Bendamustine has been used both as sole therapy and in combination with other agents including etoposide, fludarabine,mitoxantrone, methotrexate, prednisone, rituximab, vincristine and 90Y-ibritumomab tiuxetan.
Lymphomas
One combination for stage III/IV relapsed or refractory indolent lymphomas and mantle cell lymphoma (MCL), with or without prior rituximab-containing chemoimmunotherapy treatment, is bendamustine with mitoxantrone and rituximab.[5] In Germany in 2012 it has become the first line treatment of choice for indolent lymphoma.[6] after Trial results released in June 2012 showed that it more than doubled disease progression-free survival when given along with rituximab. The combination also left patients with fewer side effects than the older R-CHOP treatment.[7]
Adverse effects
Common adverse reactions are typical for the class of nitrogen mustards, and include nausea, fatigue, vomiting, diarrhea, fever, constipation, loss of appetite, cough, headache, unintentional weight loss, difficulty breathing, rashes, and stomatitis, as well as immunosuppression, anemia, and low platelet counts. Notably, this drug has a low incidence of hair loss (alopecia) unlike most other chemotherapy drugs.[8]
……………………
http://www.google.com/patents/WO2013046223A1?cl=en
First aspect of the present invention provides an improved process for the preparation of Bendamustine hydrochloride of the formula (VI)

comprising the steps of:
a) reacting a compound of the formula (II), wherein R is Ci-C6 alkyl

with a 2-haloethanol in the presence of a base to give a compound of formula (III);

b) reacting the compound of formula (III) with a chlorinating agent to provide a compound of formula (IV);

c) hydrolyzing the compound of formula (IV) with Lithium source to give a compound of formula (V); and

d) converting the compound of formula (V) to bendamustine or bendamustine hydrochloride of Formula VI .

Reference Example- 1
Preparation of Bendamustine Hydrochloride as per Patent No. DD159877
Ethyl 4-[l-methyl-5-bis-(2-hydroxyethyl)-amino-benzimidazolyl- 2]butanoate (4, 4.305g) was added to chloroform (36mL) and agitated till clear solution is formed. The solution was cooled to 0°C. Thionyl chloride (2.175g) was added to the above solution within 40 minutes maintaining the temperature of the solution to 0-5°C by cooling. The reaction mixture was agitated at 0-5°C for 1 hour. The temperature was raised slowly to room temperature by removing cooling within 2.5 to 3 hrs and subsequently agitated at room temperature for 15 to 16 hrs. The solution was dispersed by agitating in 37.5mL concentrated hydrochloric acid whereby the excessive thionyl chloride was decomposed under increased hydrochloric acid and S02development. The chloroform was distilled away and further stirred for 3 hrs at around 95°C. Activated carbon (0.78g) was added to the solution and stirred for further 30 minutes at around 95 °C. The solution was concentrated to almost 8mL under vacuum and the residue was diluted with 24mL of water and stirred up to crystallization. The further purification was done by recrystallization from water.
Example-4
Preparation of Bendamustine hydrochloride (VI) through Lithium 4-[l-methyl-5- bis-(2-chloroethyl)-benzimidazoIyl-2] butanoate (V)
Activated charcoal (11. Og) was added to Cone. HC1 (165.0 mL) under stirring and cooled to 5-10°C. Lithium 4-[l-methyl-5-bis-(2-chloroethyl)- benzimidazolyl-2] butanoate (V, HO.Og, 0.302 mol) was added below 65°C under agitation and agitated for 30-45 minutes. The reaction mass was filtered on celite bed prewashed with cone. HC1 and the celite bed was washed with cone. HC1 (27.5mL). The filtrate and washings were combined. DM water (550.0mL) was added to combined filtrate and washings and agitated for 15 minutes. DM water (1.1L) was added and stirred at 20-30°C for 30 minutes. The resulting mass was cooled to 0-5°C and maintained at a temperature of 0 to 5°C for 30 minutes under agitation. The solid was filtered, washed with chilled (0-5°C) DM water twice (220.0 mL each X 2 = 440.0mL) followed by with chilled acetone (0-5°C) (55. OmL) and sucked dried for 1 hour. The solid cake was agitated with acetone (1 lOO.OmL) for 10 minutes and filtered. The solid material was dried at 20-25°C under 100-200 mbar vacuum for one hour till moisture content is between 4.4-6.0% w/w to give the title compound (VI, 80.0g; 67.10%), with a purity of 99.86%.
…………………………..

Gao, L.; Wang, Y.; Song, D. Chinese J. New Drugs 2007, 16, 1960
Ozegowski, V. W.; Krebs, D. J. Prakt. Chem. 1963, 20, 178
Werner, W.; Letsch, G.; Ihn, W.; Sohr, R.; Preiss, R. Pharmazie 1991, 46, 113
Ozegowski, W.; Krebs, D. J. Prakt. Chem. 1963, 20, 178
Werner, W.; Letsch, G.; Ihn, W. Pharmazie 1987,42, 272
………………………………..

-
(a) Chen, J., Przyuski, K., and Roemmele, R. U.S. Patent 8,420,829, April 16, 2013;
Chem. Abstr. 2010, 152, 454105.
(b) Chen, J.; Przyuski, K.; Roemmele, R.; Bakale, R. P.Org. Process Res. Dev. 2011, 15, 1063………………………………………Org. Process Res. Dev., 2011, 15 (5), pp 1063–1072DOI: 10.1021/op200176f Process Research and Development activities leading to a new and efficient route to bendamustine hydrochloride, 1, the active ingredient in Treanda, a treatment for blood cancers, are disclosed. Two key features of this new process include a one-pot hydrogenation/dehydration sequence to construct the benzimidazole moiety and a novel reductive alkylation using chloroacetic acid and borane to install the bischloroethyl side chain. The number of synthetic steps has been significantly reduced to five from the eight in the current commercial process. The overall yield has been improved from 12% to 45%. Additionally, this new route eliminates chloroform, ethylene oxide, and sodium sulfide. Scale-up of the new route has been successfully demonstrated to prepare kilogram quantities of bendamustine hydrochloride.…………………………Org. Process Res. Dev., 2011, 15 (5), pp 1063–1072DOI: 10.1021/op200176f
Process Research and Development activities leading to a new and efficient route to bendamustine hydrochloride, 1, the active ingredient in Treanda, a treatment for blood cancers, are disclosed. Two key features of this new process include a one-pot hydrogenation/dehydration sequence to construct the benzimidazole moiety and a novel reductive alkylation using chloroacetic acid and borane to install the bischloroethyl side chain. The number of synthetic steps has been significantly reduced to five from the eight in the current commercial process. The overall yield has been improved from 12% to 45%. Additionally, this new route eliminates chloroform, ethylene oxide, and sodium sulfide. Scale-up of the new route has been successfully demonstrated to prepare kilogram quantities of bendamustine hydrochloride.…………………………Org. Process Res. Dev., 2011, 15 (5), pp 1063–1072DOI: 10.1021/op200176fPreparation of Bendamustine Hydrochloride (1)
A……../…………..purity of 99.9 A%.1H NMR (400 MHz, DMSO-d6) δ 12.3 (br s, 1H), 7.72 (d, J = 9.3 Hz, 1H), 7.14 (d, J = 2.3 Hz, 1H), 6.89 (dd, J = 9.3, 2.3 Hz, 1H), 3.90 (s, 3H), 3.80 (m, 8H), 3.14 (t, J = 7.6 Hz, 2H), 2.42 (t, J = 7.2 Hz, 2H), 2.01 (quint, J = 7.6 Hz, 2H);LC/MS (ESI, m/z) 358.2 Da (M + 1).……………………..Bendamustine, 4-[5-[bis(2-chloroethyl)amino]-l-methyl-2-benzimidazolyl]butyric acid of formula (1)

, is a cytostatic agent currently approved, in a form of a hydrochloride salt, for treatment of various cancer diseases, e.g. chronic lymphocytic leukemia. It is marketed in the form of a lyophilized powder for intravenous injection, e.g., under the brand name Ribomustin.
Bendamustine, including bendamustine hydrochloride, was first disclosed in DD 34727. Bendamustine hydrochloride may exist, in solid state, in various polymorphic forms, which are disclosed, e.g. in WO 2009/120386. The hydrochloride product disclosed in DD 34727 is a monohydrate. The original process for making bendamustine in DD 34727 comprises the following synthetic pathway:

The group R in the above process is an ethyl group.
The last step of the above process was subsequently technologically improved in DD 159877.
Without providing any experimental detail, DD 34727 also teaches that the starting compound of formula (4) for the above process may be prepared from 2-methylamino-5-nitro- aniline of formula (2) and glutaric acid anhydride. The obtained anilide of formula (3) is cyclized in diluted hydrochloric acid.

Li-Mei et al, in Zhongguo Xinyao Zazhi, Chinese Journal of New Drugs (2007), 16(23), 1960-1961, disclose a process for the preparation of bendamustine hydrochloride in a total yield of 33.5%, which also involves reacting the compound of formula (10) with ethylene oxide to give compound (11). Starting from 2,4-dinitro-l-chlorobenzene, compound (11) is obtained in an overall yield of about 40%.IP.com Journal 2009, 9(7B), 21 discloses a process for the preparation of ethyl-4-[5-[bis(2- hydroxyethyl)amino]-l-methylbenzimidazol-2-yl]butanoate (11) [R=Et], wherein the corresponding compound of formula (10) reacts, instead of ethylene oxide, with 2-halo ethanol in the presence of an inorganic base.
A similar process has been disclosed in WO 2011/079193, wherein the base employed in the reaction of the compound of formula (10) with the 2-haloethanol is an organic base, which is advantageous over inorganic base. The preferred ester group R in the compounds (10) and (11) is the 2-propyl group.
WO 2010/042568 discloses a second basic process for making bendamustine, which is based on providing the compound of formula (5)

(5)
, wherein R is typically a methyl group, by a two step synthesis starting from 2,4- dinitroaniline of formula (6) via the dinitroanilide of formula (7)

This compound of formula (5) is subjected, at reductive conditions (preferably hydrogenation over a platinum catalyst), to a cyclization reaction forming a compound of formula (8)

(8)
, which subsequently may be dehydrated by a strong acid to yield the compound of formula
(10) above. The substituent R in both formulas is a methyl group.
The compound of formula (10) is advantageously subjected to a reductive alkylation with a chloroacetic acid or chloroacetylaldehyde. The reductive agent in the alkylation is suitably a borane or a borohydride. This way, the bendamustine ester of formula (la)

, wherein R is a methyl group, is made directly, without need of forming an intermediate bis-hydroxyethyl compound (11). In the last step of the overall process, the ester (la) is hydrolyzed by a strong acid.
In any process of making bendamustine, various impurities are formed due to various reactive groups in the molecule.
The subject of the present invention is a novel synthetic route to intermediates involved in the synthesis of bendamustine of formula (1) as well as of salts and esters thereof. The approach is based on a novel use of a compound of formula (13) below as the starting material in a synthetic transformation leading to bendamustine, or a pharmaceutically acceptable salt thereof.
In a first aspect, the invention provides a process for making a compound of formula (11), or a salt thereof,

wherein R is hydrogen or a C1-C4 alkyl group,
said process comprising the following steps:
a] providing the compound of formula (13), preferably by reaction of the compound of formula (12) with methylamine,

(12) (13)b] reduction of the compound of formula (13), preferably by hydrogen under catalysis by a transition metal, to an amino compound of formula (14),

c] condensation of the compound of formula (14) with glutaric acid anhydride, or a functional analogue thereof, providing a tertiary alcohol compound of formula (15)

and/or any tautomeric forms thereof according to formula (14A) or (14B)

(14A), (14B),
d] dehydratation and, optionally, esterification of the product of the step c) , preferably in the presence of a strong acid, to yield the compound of formula (11).
In a particular aspect, the above process sequence leading to a compound of formula (11) further comprises a subsequent step of converting the compound of formula (11) to
bendamustine, a salt thereof or an ester thereof, conventionally by reaction with thionyl chloride, followed by ester hydrolysis and salt formation using hydrochloric acid. In yet another aspect, the process sequence of the above steps a) to c) or, optionally, of the above steps a) to d), is performed without isolation or purification of intermediates.
The compounds of formula (14), (14A), (14B) and (15), the above processes of making them, and the use thereof as a starting material for making compounds of formula (11) and/or bendamustine of formula (1), or a pharmaceutically acceptable salt thereof, form next particular aspects of the present invention.

Example 3
A solution of [11, R = Me] (4.0 g, 12 mmol) in dichloromethane (40.0ml) was prepared.
A 100 ml, three -necked, round -bottomed flask equipped with a magnetic stirring bar and a reflux condenser was charged with thionyl chloride (3.60 g, 2.20 ml, 30 mmol) and dichloromethane (12.0 ml) to produce a clear solution. The latter was stirred at 500 rpm at 23 °C and the solution of [11 , R = Me] was added over 15 min via a syringe pump. The resulting mixture was stirred at 500 rpm at 23 °C for 15 min and then at 35 °C for 3.0 h. An aqueous solution of hydrochloric acid (19.4 %) was prepared by mixing of concentrated hydrochloric acid (4.7 g, 4.0 ml) with water (4.0 g, 4.0 ml) and the solution was charged to the reaction mixture. The mixture was further stirred at 500 rpm at 60 °C for 2.0 h under reduced pressure of 100 mbar and the escaping volatiles were condensed and collected outside the reaction vessel. An aqueous solution of hydrochloric acid (4.0 ml, 5 M) was charged in order to dilute the reaction mixture. The mixture was filtered through diatomaceous earth and the filter cake was washed with aqueous solution of hydrochloric acid (2x 1.0 ml, 5 M). The collected filtrate was treated with activated carbon, the used carbon was filtered off, washed with aqueous solution of hydrochloric acid (2x 1.0 ml, 5M), and the filtrate was collected to a 100 ml, round -bottomed flask equipped with a magnetic stirring bar. The filtrate was stirred and diluted with water (40.0 g, 40.0 ml). The slurry was filtered and the filter cake was washed with water (2x 1.0 ml). The filter cake (3.8 g) was charged to a 25 ml, round-bottomed flask equipped with a magnetic stirring bar containing an aqueous solution of hydrochloric acid (8.5 ml, 5M). The mixture was stirred at 60 °C until the solids were dissolved and activated carbon (0.38 g) was charged. The mixture was stirred at 40 °C for additional 5 min and the suspension was filtered through a diatomaceous earth pad. The filter cake was washed with aqueous solution of hydrochloric acid (2x 1.0 ml, 5M) and the filtrate was collected to a 100 ml, round -bottomed equipped with a magnetic stirring bar. The filtrate was stirred at 23 °C and water (42.0 g, 42.0 ml) was added and the mixture was stirred at 23 °C for additional 1 h. The slurry of crystals was filtered and the filter cake was washed with aqueous solution of hydrochloric acid (2x 3.0 ml, 5M). The filtrate was discarded and the filter cake was dried to produce Bendamustine hydrochloride monohydrate (3.3 g) with an overall isolated yield of 67 % and with a chemical purity of 99.9 % by HPLC peak area normalization.
Characteriz ation
lU NMR (400 MHz, DMSO-J<5): δ (ppm) = 2.05 (q, J = 7.51 Hz, 2H), 2.41 (t, J = 7.19 Hz, 2H), 3.18 (t, / = 7.63 Hz, 2H), 3.79 (m, 8H), 3.90 (s, 3H), 6.95 (d, J = 2.31 Hz, 1H), 7.11 (dd, Jx = 2.40 Hz, J2 = 9.20 Hz, 1H), 7.80 (d, J = 9.20 Hz, 1H).
Assay H20 (Karl-Fisher titration): 4.7 %
Assay HC1 (argentometric titration): 8.8 %
Bendamustine 
Systematic (IUPAC) name 4-[5-[Bis(2-chloroethyl)amino]-1-methylbenzimidazol-2-yl]butanoic acid Clinical data Trade names Treanda AHFS/Drugs.com Consumer Drug Information MedlinePlus a608034 Licence data US FDA:link Pregnancy cat. - US: D
Legal status Routes Intravenous infusion Pharmacokinetic data Bioavailability NA (intravenous only) Protein binding 94–96% Metabolism Hydrolyzed to inactive metabolites. Two minor metabolites (M3 and M4) formed by CYP1A2 Half-life 40 min (bendamustine), 3 h (M3), 30 min (M4) Excretion Mostly fecal Identifiers CAS number 16506-27-7 
ATC code L01AA09 PubChem CID 65628 ChemSpider 59069 UNII 9266D9P3PQ ChEMBL CHEMBL487253 Chemical data Formula C16H21Cl2N3O2 Mol. mass 358.262 g/mol References
- Kath R, Blumenstengel K, Fricke HJ, Höffken K (January 2001). “Bendamustine monotherapy in advanced and refractory chronic lymphocytic leukemia”. J. Cancer Res. Clin. Oncol. 127(1): 48–54. doi:10.1007/s004320000180. PMID 11206271.
- Bagchi S (August 2007). “Bendamustine for advanced sarcoma”. Lancet Oncol. 8 (8): 674. doi:10.1016/S1470-2045(07)70225-5. PMID 17726779.
- “Cephalon press release – Cephalon Receives FDA Approval for TREANDA, a Novel Chemotherapy for Chronic Lymphocytic Leukemia”. Retrieved 2008-03-23.
- “Cephalon press release -Cephalon Receives FDA Approval for TREANDA to Treat Patients with Relapsed Indolent Non-Hodgkin’s Lymphoma”. Retrieved 2008-11-03.
- Weide R, Hess G, Köppler H, et al. (2007). “High anti–lymphoma activity of bendamustine/mitoxantrone/rituximab in rituximab pretreated relapsed or refractory indolent lymphomas and mantle cell lymphomas. A muticenter phase II study of the German Low Grade Lymphoma Study Group (GLSG)”. Leuk. Lymphoma. 48 (7): 1299–1306. doi:10.1080/10428190701361828.PMID 17613757.
- New Combo Replaces CHOP for Lymphoma. Dec 2012
- “‘Rediscovered’ Lymphoma Drug Helps Double Survival: Study”. June 3, 2012.
- Tageja, Nishant; Nagi, Jasdeepa; “Bendamustine: something old, something new”; Cancer Chemotherapy and Pharmacology, 2010 Aug;66(3):413-23. doi: 10.1007/s00280-010-1317-x.
External links
- Manufacturer’s official website intended for US patients
References:Bifunctional alkylating agent. Prepn: W. Ozegowski, D. Krebs, J. Prakt. Chem. 20, 178 (1963); eidem,Zentralbl. Pharm. Pharmakother. Laboratoriumsdiagn. 110, 1013 (1971). Antitumor activity: W. Jungstand et al., ibid. 1021.Capillary GC determn in plasma: H. Weber et al., J. Chromatogr. 525, 459 (1990).Toxicity study: U. Horn et al., Arch. Toxicol.Suppl. 8, 504 (1985).Clinical evaluation in non-Hodgkin’s lymphomas: K. Bremer, J. Cancer Res. Clin. Oncol. 128, 603 (2002); in chronic lymphocytic leukemia: T. Lissitchkov et al., ibid. 132, 99 (2006); with prednisone in multiple myeloma: W. Pönisch et al.,ibid 205.Review of pharmacology and clinical development: K. Bremer, W. Roth, Tumordiagn. Ther. 17, 1-6 (1996); J. A. Barman Balfour, K. L. Goa, Drugs 61, 631-638 (2001).WO2009120386A2 Mar 26, 2009 Oct 1, 2009 Cephalon, Inc. Novel solid forms of bendamustine hydrochloride WO2010036702A1 Sep 23, 2009 Apr 1, 2010 Cephalon, Inc. Liquid formulations of bendamustine WO2010042568A1 Oct 7, 2009 Apr 15, 2010 Cephalon, Inc. Processes for the preparation of bendamustine WO2011079193A2 Dec 22, 2010 Jun 30, 2011 Dr. Reddy’s Laboratories Ltd. Preparation of bendamustine and its salts DD34727A Title not available DD159877A1 Title not available US20060128777 Nov 4, 2005 Jun 15, 2006 Bendall Heather H Cancer treatments US20060159713 Jan 12, 2006 Jul 20, 2006 Cephalon, Inc. Bendamustine pharmaceutical compositions













Ramatroban

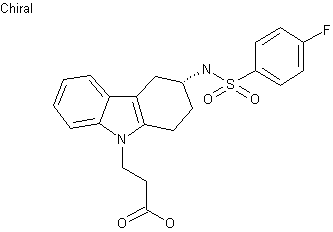

| Formula |
C21H21FN2O4S
|
|---|---|
| CAS |
116649-85-5
|
| Mol weight |
416.4658
|
- 3-(4-fluorophenylsulfonamido)-1,2,3,4-tetrahydro-9-carbazole propanoic acid
- BAY u 3405
- BAY u 3406
- BAY u-3405
- BAY u3405
- ramatroban
Ramatroban (INN) (also known as Bay-u3405)[1] is a thromboxane receptor antagonist.[2]
It is also a DP2 receptor antagonist.[3]
It is indicated for the treatment of coronary artery disease.[4] It has also been used for the treatment of asthma.[5]
It was developed by the German pharmaceutical company Bayer AG and is co-marketed in Japan by Bayer and Nippon Shinyaku Co. Ltd. under the trade name Baynas.
SYN
Science 1976,193163-5
Proc Natl Acad Sci USA 1975,72(8),2994-8
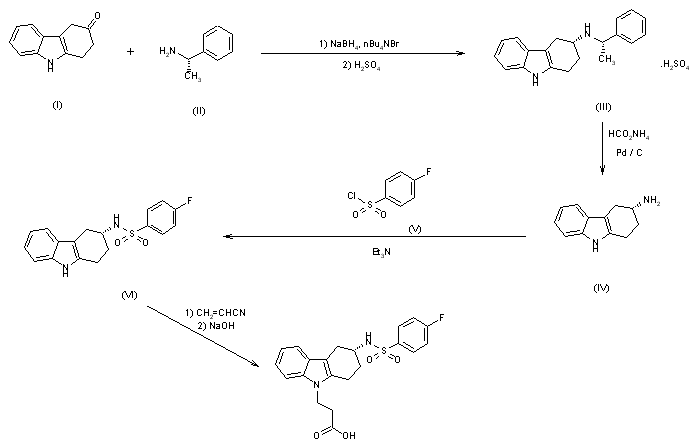
The synthesis of Bay u 3405 was carried out as follows: Reductive amination of 3-oxo-1,2,3,4-tetrahydrocarbazole (I) with S-phenethylamine (II) afforded a mixture of diastereomeric amines, of which the desired isomer (III) crystallized in high diastereomeric purity as the hydrogensulfate. Cleavage of the phenethyl group by transfer hydrogenolysis with amminium formate and palladium on charcoal yielded the enantiomerically pure (3R)-3-amino-1,2,3,4-tetrahydrocarbazole (IV). Sulfonylation of (IV) with 4-fluorobenzenesulfonyl chloride (V) to the sulfonamide (VI) followed by addition of acrylonitrile and subsequent hydrolysis gave Bay u 3405.
SYN
J Label Compd Radiopharm 1994,34(12),1207
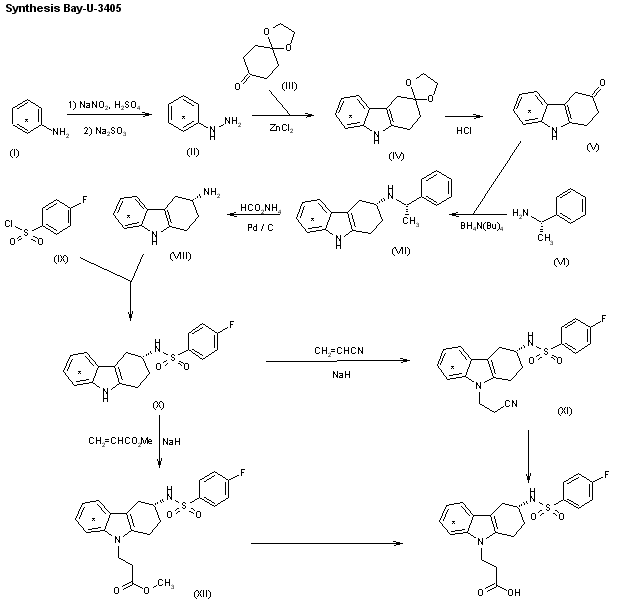
The synthesis of [14C]-labeled Bay-u-3405 by two closely related ways has been described: 1) [14C]-Labeled aniline (I) is diazotized and reduced with sodium sulfite, yielding the labeled hydrazine (II), which is condensed with the monoketal of cyclohexane-1,4-dione (III) under Fisher’s indole synthesis (ZnCl2) to afford the tetrahydrocarbazole (IV). The hydrolysis of (IV) with HCl in THF/water yields 1,2,3,4-tetrahydrocarbazol-3-one (V), which is submitted to a reductive condensation with (S)-1-phenylethylamine (VI) by means of tetrabutylammonium borohydride, yielding preferentially the secondary amine (VII), which, after purification, is dealkylated with ammonium formate and Pd/C to afford 1,2,3,4-tetrahydrocarbazole-3(R)-amine (VIII). The acylation of (VIII) with 4-fluorophenylsulfonyl chloride (IX) gives the corresponding sulfonamide (X), which is condensed with acrylonitrile by means of NaH, yielding 3-[3(R)-(4-fluorophenylsulfonamido)-1,2,3,4-tetrahydrocarbazol-9-yl]pro pionitrile (XI). Finally, this compound is hydrolyzed in the usual way. 2) The condensation of the sulfonamide (X) with methyl acrylate by means of NaH as before gives 3-[3(R)-(4-fluorophenylsulfonamido)-1,2,3,4-tetrahydrocarbazol-9-yl]propionic acid methyl ester (XII), which is finally hydrolyzed in the usual way.
……………………

http://pubs.rsc.org/en/content/articlelanding/2012/oc/c2oc90018a#!divAbstract
………………….
http://onlinelibrary.wiley.com/doi/10.1002/adsc.201300993/abstract












135−137 °C; IR (KBr) ν 3276, 2926, 1712, 1591, 1494, 1467, 1153
cm−1
; 1
H NMR (CD3OD, 300.13 MHz) δ 1.87−2.09 (m, 2H14),
2.47−2.54 (m, 1H11), 2.67 (t, 3
JHH = 6.7 Hz, 2H2), 2.75−2.91 (m,
2H13+1H11), 3.61−3.71 (m, 1H12), 4.30 (t, 3
JHH = 6.7 Hz, 2H3), 6.96
(t, 3
JHH = 6.9 Hz, 1H7), 7.08 (t, 3
JHH = 7.1 Hz, 1H6), 7.21−7.31 (m,
2H18+H5+H7), 7.93−7.98 (m, 2H17); 13C NMR (CD3OD, 75.5 MHz)
δ 21.2 (C14), 29.7 (C11), 31.1 (C13), 35.8 (C2), 40.0 (C3), 51.5 (C12),
108.0 (C10), 110.2 (C5), 117.4 (d, 2
JCF= 23.3 Hz, 2C18), 118.7 (C8),
120.2 (C7), 122.4 (C6), 128.9 (C19), 131.0 (d, 3
JCF= 9.7 Hz, 2C17),
135.3 (C15), 138.0 (C4), 139.8 (d, 4
JCF= 3.5 Hz, C16), 166.6 (d, 1
JCF=
251.4 Hz, C19), 175.2 (C1); HRMS (ESI+, m/z) calcd for
(C21H22FN2O4S)+ (M + H)+ 417.1279, found 417.1273; [α]D
20=
+64.4 (c 1, MeOH) for 99% ee.
This invention relates to 2-amino- tetrahydrocarbazole-propanoic acid and a new process for its synthesis .
2-Amino-tetrahydrocarbazole-propanoic acid is a key intermediate for the synthesis of Ramatroban, a thromboxaneA2 receptor (TP) antagonist with clinical efficacy in asthma and allergic rhinitis.

Ramatroban l-Amino-tetrahydrocarbazole-proanoic acid
US Patent 4988820 discloses the synthesis of this compound stating from compound 1, which is condensed with phenylhydrazine and ring-closed to give indole 2. Deprotection of 2 using acid provides ketone 3. Reductive amination of ketone with s-phenylethylamine in the presence of tetrabutylammonium borohydride provides compound 4, which undergoes palladium catalyzed hydrogenation to give key intermediate 5.

Ramatroban

The process, however, has disadvantages: the starting material 1 is relatively expensive, and the yield of the amination step is only 40% and needs expensive tetrabutylammonium borohydride as the reducing agent. And also the subsequent hydrogenation provides only 70% of the desired compound 5. [0006] US Patent 4988820 also describes an alternative synthesis of compound 5 starting from compound 6, which is oxidized by chromium trioxide to afford ketone 7. Condensation of compound 7 with phenylhydrazine and ring closure give indole 8. The subsequent hydrolysis using HCl provides indole 9. The intermediate 5 is obtained by resolution of racemic 9 using ( + ) -mandelic acid as the resolving agent.

9 5
However, this process has crucial disadvantages: the first step oxidation reaction needs the heavy metal reagent chromium trioxide, which is toxic and expensive, and the resolution of indole 9 using (+) -mandelic acid affords only -10 % of compound 5.
US Patent 5684158 discloses the synthesis of 2- amino-tetrahydrocarbazole-propanoic acid ethyl ester 10 by the alkylation of compound 5 in the presence of about 1 mol of alkali metal hydroxides and phase-transfer catalysts such as potassium hydroxide and benzyltriethylammonium chloride.

The problem with this reaction is that the insoluble material in the reaction mixture becomes very sticky during the reaction. The reaction mixture must be filtered in hot solvent in order to remove insoluble material during work up and the sticky material tents to block the filtration. [0010] Therefore, there is a great need for a new process for the synthesis of 2-amino-tetrahydrocarbazole- propanoic acid.
-
9- (2-carboxyethyl) -4- (4-fluorphenylsulfonamidomethyl) -1,2,3,4-tetrahydrocarbazole

-
0.91 g 9-(2-Cyanoethyl)-4-[N-(4-fluorphenylsulfonyl)-N-(2-cyanoethyl)aminomethyl]-1,2,3,4-tetrahydrocarbazol be hydrolyzed analogously to Example 7. One obtains 0.77 g (89% of theory) of crystalline product as the sodium salt.
-
M.p .: 160 ° CR f = 0.57 CH 2 Cl 2: CH 3 0H = 9: 1

-
-
(+) – 3- (4-fluorophenylsulphonamido) -9- (2-carboxyethyl) -1,2,3,4-tetrahydrocarbazole

-
5.8 g (0.0128 mol) of Example 67 are dissolved in 60 ml isopropanol, treated with 130 ml of 10% potassium hydroxide solution, after 16 hours heating under reflux, is cooled, diluted with water and extracted with ethyl acetate. The aqueous phase is concentrated in vacuo and then treated dropwise with vigorous stirring with conc.Hydrochloric acid. The case precipitated acid is filtered off, washed with water and dried thoroughly in vacuo.Obtained 4.4 g (86.6% of theory) of the product. .: Mp 85-95 ° C rotation [α] 20 = 42.55 ° (CHCl 3) D
-

Example 70
-
(-) – 3- (4-fluorophenylsulphonamido) -9- (2-tarboxyethyl) -1,2,3,4-tetrahydrocarbazole

-
The preparation of Example 70 from Example 68 is carried out analogously to the preparation of Example 69 from Example 67. m.p .: 85-95 ° C optical rotation: [α] 20 = -37.83 ° (CHCl 3) D

Synthesis pathway
| Synthesis of a) |
|---|
     |
Trade names
| Country | Trade name | Manufacturer |
|---|---|---|
| Japan | Baynas | Bayer |
| Ukraine | no | no |
Formulations
-
50 mg tablet 75 mg
Reference
-
DE 3631824 (Bayer AG; appl. 19.9.1986; prior. 21.2.1986).
-
EP 728 743 (Bayer AG; appl. 14.2.1996; D-prior. 27.2.1995).
| Patent | Submitted | Granted |
|---|---|---|
| Phenylsulfonamid substituted pyridinealken- and aminooxyalkan-carboxylic-acid derivatives. [EP0471259] | 1992-02-19 | 1995-05-17 |
| Heterocyclic substituted cycloalkano(b)-indolesulfonamides. [EP0473024] | 1992-03-04 | |
| Cycloalkano[b]dihydroindoles and -indolesulphonamides substituted by heterocycles. [EP0451634] | 1991-10-16 | 1994-03-09 |
| Respiratory Drug Condensation Aerosols and Methods of Making and Using Them [US2009258075] | 2009-10-15 | |
| ANTITHROMBOTIC SUBSTITUTED CYCLOALKANO(B)DIHYDROINDOLE- AND -INDOLE-SULPHONAMIDES [US5096897] | 1992-03-17 | |
| Indolesulphonamide-substituted dihydropyridines [US5272161] | 1993-12-21 | |
| THERMODYNAMICALLY STABLE FORM OF (R)-3-[ [(4-FLUOROPHENYL) SULPHONYL]AMINO] -1,2,3,4- TETRAHYDRO -9H-CARBAZOLE -9-PROPANOIC ACID (RAMATROBAN) [WO9933803] | 1999-07-08 |
| DE1695703B2 * | Mar 15, 1967 | Nov 20, 1975 | Sumitomo Chemical Co., Ltd., Osaka (Japan) | Title not available |
| DE2125926A1 * | May 25, 1971 | Jan 27, 1972 | Title not available | |
| DE2226702A1 * | May 25, 1972 | Dec 13, 1973 | Schering Ag | Neue mittel zur behandlung des diabetes mellitus |
| FR1415322A * | Title not available | |||
| GB1487989A * | Title not available | |||
| US4235901 * | May 14, 1979 | Nov 25, 1980 | American Home Products Corporation | 1-Hydroxyalkanamine pyrano(3,4-b)indole compositions and use thereof |
-
References
- ^ “Ramatroban (compound)”. PubChem. National Center for Biotechnology Information. Retrieved 22 June 2019.
- ^ Sugimoto H, Shichijo M, Iino T, et al. (April 2003). “An orally bioavailable small molecule antagonist of CRTH2, ramatroban (BAY u3405), inhibits prostaglandin D2-induced eosinophil migration in vitro”. J. Pharmacol. Exp. Ther. 305 (1): 347–52. doi:10.1124/jpet.102.046748. PMID 12649388.
- ^ Royer JF, Schratl P, Carrillo JJ, et al. (September 2008). “A novel antagonist of prostaglandin D2 blocks the locomotion of eosinophils and basophils”. Eur. J. Clin. Invest. 38 (9): 663–71. doi:10.1111/j.1365-2362.2008.01989.x. PMID 18837743.
- ^ Fiedler VB, Seuter F, Perzborn E (December 1990). “Effects of the novel thromboxane antagonist Bay U 3405 on experimental coronary artery disease” (PDF). Stroke. 21 (12 Suppl): IV149–51. PMID 2260140.
- ^ Endo S, Akiyama K (November 1996). “[Thromboxane A2 receptor antagonist in asthma therapy]”. Nippon Rinsho (in Japanese). 54 (11): 3045–8. PMID 8950952.
External links
- (in Japanese) Baynas Tablets Prescribing Information
 |
|
| Systematic (IUPAC) name | |
|---|---|
| 3-((3R)-3-{[(4-fluorophenyl)sulfonyl]amino}-1,2,3,4-tetrahydro-9H-carbazol-9-yl)propanoic acid | |
| Clinical data | |
| Legal status |
|
| Routes | Oral |
| Identifiers | |
| CAS number | 116649-85-5 |
| ATC code | None |
| PubChem | CID 123879 |
| IUPHAR ligand | 1910 |
| ChemSpider | 110413 |
| UNII | P1ALI72U6C |
| ChEMBL | CHEMBL361812 |
| Chemical data | |
| Formula | C21H21FN2O4S |
| Mol. mass | 416.46 g/mol |
 |
|
| Clinical data | |
|---|---|
| Trade names | Baynas |
| AHFS/Drugs.com | International Drug Names |
| Routes of administration |
Oral (tablets) |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.159.668 |
| Chemical and physical data | |
| Formula | C21H21FN2O4S |
| Molar mass | 416.47 g·mol−1 |
| 3D model (JSmol) | |

NEW DRUG APPROVALS
ONE TIME
$10.00
A Flow Reactor with Inline Analytics: Design and Implementation

A Flow Reactor with Inline Analytics: Design and Implementation
-
Wu, H., Dong, Z., Haitao, L., and Khan, M. Org. Process Res. Dev. 2014, 18, 10.1021/op500056a.
-
(a) Razzaq, T.; Kappe, C. O. Chem.—Asian J. 2010, 5, 1274– 1289
(b) Wiles, C.; Watts, P. Green Chem. 2014, 16, 55– 62
(c) Moseley, J. D.; Woodman, E. K. Org. Process Res. Dev. 2008, 12, 967– 981
-
Ullah, F.; Samarakoon, T.; Rolfe, A.; Kurtz, R. D.; Hanson, P. R.; Organ, M. G. Chem.—Eur. J. 2010, 16, 10959– 10962
and references cited therein
-
Guidance for Industry, Q8(R2) Pharmaceutical Development; U. S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research, Center for Biologics Evaluation and Research: Silver Springs, MD, 2009; p 18.
-
Calibrese, G. S.; Pissavini, S. AIChE J. 2011, 54, 828– 834
-
(a) Alsten, J. G.; Reeder, L. M.; Stanchina, C. L.; Knoechel, D. J. Org. Process Res. Dev. 2008, 12, 989– 994
(b) Roberge, D. M.; Zimmermann, B.; Rainonee, F.; Gottsponer, M.; Eyholzer, M.; Kockmann, N. Org. Process Res. Dev. 2008, 12, 905– 910

Vijay Kirpalani
vk@pi-inc.co
CEO
Pi-inc (Process Intensification Experts LLP)
READ AT….https://newdrugapprovals.org/2014/11/11/flow-chemistry-test-facility-in-india/
Toronto, canada

2-aminooctahydrocyclopentalene-3a-carboxamides as potent CCR2 antagonists

((2R,3aR,6aR)-2-((3R,4R)-3-Methoxytetrahydro-2H-pyran-4-ylamino)octahydropentalen-3a-yl)(3-(trifluoromethyl)-7,8-dihydro-1,6-naphthyridin-6(5H)-yl)methanone
((2R,3aR,6aR)-2-((3R,4R)-3-Methoxytetrahydro-2H-pyran-4-ylamino)octahydropentalen-3a-yl)(3-(trifluoromethyl)-7,8-dihydro-1,6-naphthyridin-6(5H)-yl)methanone Semisuccinate
D-erythro-Pentitol, 1,5-anhydro-3,4-dideoxy-3-[[(2R,3aR,6aR)-3a-[[7,8-dihydro-3-(trifluoromethyl)-1,6-naphthyridin-6(5H)-yl]carbonyl]octahydro-2-pentalenyl]amino]-2-O-methyl-, rel–
cas 1416178-01-2
Abbott Laboratories, Abbott Laboratories Trading (Shanghai) Company, Ltd.
Antagonists of the CC-chemokine receptor 2 (CCR-2) have been vigorously pursued by a number of pharmaceutical companies as a target for drug discovery, in that compounds could have the potential for use in the acute and chronic conditions of inflammatory and autoimmune diseases associated with infiltration of monocytes, macrophages, lymphocytes, dendritic cells, NK cells, eosinophils, basophils, natural killer (NK cells), and memory T-cells. A compound of interest that was discovered in the Janssen laboratories that met the initial criteria set out during the in vitro screening phase of drug discovery was bicyclic 1
Chemokines are chemotactic cytokines that are released by a wide variety of cells to attract leukocytes, as illustrated by macrophages, T cells, B cells, eosinophils, basophils, and neutrophils to and from sites of inflammation or within specific compartments, as illustrated by lymph nodes (reviewed in Schall, Cytokine 1991 ; 3: 165- 183; Schall, et al., Curr. Opin. Immunol. 1994; 6:865- 873; and Murphy, Rev. Immun. 1994; 12:593-633). In addition to stimulating chemotaxis, other changes can be selectively induced by chemokines in responsive cells, including changes in cell shape, transient rises in the concentration of intracellular free calcium ions ([Ca2+]), granule exocytosis, integrin upregulation, formation of bioactive lipids (e.g., leukotrienes), and respiratory burst, associated with leukocyte activation. Thus, the chemokines are early modulators of inflammatory response, effecting inflammatory mediator release, chemotaxis and extravasation to sites of infection or inflammation.
There are four classes of chemokines, CXC (a), CC (β), C (γ), and CX3C (δ), depending on whether the first two cysteines are separated by a single amino acid (C-X- C), are adjacent (C-C), have a missing cysteine pair (C), or are separated by three amino acids (CX3C). The a-chemokines, such as interleukin-8 (IL-8), melanoma growth stimulatory activity protein (MGSA), and stromal cell derived factor 1 (SDF-1) are chemotactic primarily for neutrophils and lymphocytes, whereas β-chemokines, such as RANTES, ΜΙΡ-Ια, ΜΙΡ- Ι β, monocyte chemotactic protein- 1 (MCP-1), MCP-2, MCP-3, and eotaxin are chemotactic for macrophages, T-cells, eosinophils and basophils (Deng, et al., Naturel996; 381 :661-666). The C chemokine lymphotactin shows specificity for lymphocytes (Kelner, et al., Science 1994; 266: 1395-1399) while the CX3C chemokine fractalkine shows specificity for lymphocytes and monocytes (Bazan, et al., Nature 1997; 385:640-644).
Chemokines bind specific cell-surface receptors belonging to the family of G- protein-coupled seven-transmembrane-domain proteins (reviewed in Horuk, Trends
Pharm. Sci. 1994; 15: 159-165) termed “chemokine receptors.” On binding their cognate ligands, chemokine receptors transduce an intracellular signal through the associated heterotrimeric G protein, resulting in a rapid increase in intracellular calcium concentration. There are at least twelve human chemokine receptors that bind or respond to β-chemokines with the following characteristic pattern: CCR1 (or “CKR-1 ” or “CC- CKR-1 “) ΜΙΡ-Ια, ΜΙΡ-Ι β, MCP-3, RANTES (Ben-Barruch, et al., J. Biol. Chem. 1995; 270:22123-22128; Neote, et al., Cell 1993; 72:415425); CCR2A and CCR2B (or “CKR- 2A’7″CKR-2A” or “CC-CKR-2A”/”CC-CKR2A”) MCP-1, MCP-2, MCP-3, MCP-4; CCR3 (or “CKR-3” or “CC-CKR-3”) eotaxin, RANTES, MCP; (Ponath, et al., J. Exp. Med. 1996; 183:2437-2448); CCR4 (or “CKR-4” or “CC-CKR-4”) TARC, MDC (Imai, et al., J. Biol. Chem. 1998; 273: 1764- 1768); CCR5 (or “CKR-5” or “CC-CKR-5”) MIP- la, RANTES, MIP-Ι β; (Sanson, et al., Biochemistry 1996; 35:3362-3367); CCR6 MIP-3a (Greaves, et al., J. Exp. Med. 1997; 186:837-844); CCR7 ΜΙΡ-3β and 6Ckine (Campbell, et al., J. Cell. Biol. 1998; 141 : 1053- 1059); CCR8 I- 309, HHV8 vMIP-I, HHV-8 vMIP-II, MCV vMCC-I (Dairaghi, et al., J. Biol. Chem. 1999; 274:21569-21574); CCR9 TECK (Zaballos, et al., J. Immunol. 1999; 162:5671-5675), D6 MIP-1 beta, RANTES, and MCP-3 (Nibbs, et al., J. Biol. Chem. 1997; 272:32078-32083), and the Duffy blood-group antigen RANTES, MCP-1 (Chaudhun, et al., J. Biol. Chem. 1994; 269:7835-7838).
Chemokine receptors, such as CCR1, CCR2, CCR3, CCR4, CCR5, CCR6, CCR7, CCR8, CCR9, CXCR1, CXCR2, CXCR3, CXCR4, CXCR5, CX3CR1, and XCRl have been implicated as being important mediators of inflammatory and immunoregulatory disorders and diseases, including asthma and allergic diseases, as well as autoimmune pathologies such as rheumatoid arthritis and atherosclerosis.
The CCR2 chemokine receptor is expressed primarily in monocytes and activated T lymphocytes, and its functional activity can be measured by cytosolic calcium elevation or chemotaxis. CCR2 exists in two isoforms, CCR2A and CCR2B. These two isoforms are alternatively spliced variants of a single MCP- 1 receptor gene and differ only in the carboxyl-terminal tails. The chromosomal location of the CCR2 gene is localized to
3p21. The CC chemokines, MCP-1, MCP-2, MCP-3, and MCP-4, have been identified as the ligands that are selective and of high affinity to the CCR2 receptor.
The highly selective expression of CCR2 makes it an ideal target for intervention to interrupt inappropriate monocyte and T cell trafficking. The clinical indications for such intervention are in inflammatory diseases and T-cell mediated autoimmune diseases such as multiple sclerosis, rheumatoid arthritis, asthma, allergy, chronic obstructive pulmonary disease, atherosclerosis, restinosis, type I and type II diabetes, metabolic syndrome, and pain. Ectopic expression of MCP-1 and CCR2 in certain tumors indicates that selective modulation (such as antagonism or inhibition) of CCR2 can have value in tumor immunotherapy, particularly attenuation of metastasis.
The native peptide ligand of CCR2 is monocyte chemoattractant protein- 1 (MCP- 1 or CCL2) containing two adjacent disulfied bonds. Ample evidence exists for the role of the CCR2/MCP-1 system in preclinical animal models of pain (White F.A., Jung F., and Miller R.J., Proc. Natl. Acad. Sci. USA 2007; 51 :20151). Although CCR2 and MCP- 1 have limited expression levels in the CNS tissues under normal conditions, significant upregulation of CCR2 and MCP- 1 has been observed following a neuropathic injury in tissue relevant to pain, including neurons and glia in the spinal cord, rostroventromedial medulla (RVM) and DRG (Wang H., Zou S., Wei F., Dubner R., and Ren K., Soc for Neurosci Poster 2009; 72.3). MCP- 1 has been shown to increase the excitability of neurons acutely dissociated from the DRG tissue (Sun J.H., Yang B., Donnelly D.F., Ma C, and LaMotte R.H., J Neurophysiol. 2006; 96:2189). In addition, direct injection of MCP- 1 in the spinal cord induces thermal hyperalgesia and mechanical allodynia (Dansereau et al. Neurochem. 2008; 106:7), and the MCP- 1 induced pronociception can be blocked by a CCR2 antagonist, INCB3344. Similarly, the hyperalgesia induced by MCP-1 injection in the RVM is reversed by another CCR2 antagonist, RS I 02895 (Wang H., Zou S., Wei F., Dubner R. and Ren K., Soc for Neurosci Poster 2009; 72.3). In addition, CCR2 knock out mice exhibit significantly reduced mechanical allydonia following nerve injury and reduced nocifensive behavior in the second phase of the formalin model, whereas they exhibit normal sensitivity to acute pain stimulation in the hot plate model (Abbadie C, Lindia J.A., Cumiskey A.M., Peterson L.B., Mudgett J.S., Bayne E.K., DeMartino J.A., Maclntyre D.E., and Forrest M.J., Proc Natl Aca Sci USA 2003; 100:7947). Treatment with AZ889 (Serrano A., Pare M., Mcintosh F., Elmes S.J.R. Martino G., Jomphe C, Lessard E., Lembo P.M.C., Vaillancourt F., Perkins M.N., and Cao C.Q., Mol. Pain 2010; 6:90), a CCR2 antagonist, abolished CCL2-evoked neuronal excitation, confirming that this activity is CCR2 -mediated. Neuronal and non-neuronal cells in the spinal cord were also excited by CCL2 applications indicating an important role of spinal CCR2 in neuropathic pain. In vivo spinal intrathecal injection of AZ889 produced dose-dependent analgesia in chronic constriction injury rats (Serrano A., Pare M., Mcintosh F., Elmes S.J.R., Martino G., Jomphe C, Lessard E., Lembo P.M.C., Vaillancourt F., Perkins M.N., and Cao C.Q., Mol. Pain 2010; 6:90). Additionally, application of AZ889 to the exposed spinal cord inhibited evoked neuronal activity and confirmed that CCR2-mediated analgesia involved predominantly the spinal cord. In view of the clinical importance of CCR2, the identification of compounds that modulate CCR2 function represents an attractive avenue into the development of new therapeutic agents that can be used to treat diseases such as inflammatory, autoimmune disease, cancer, and pain, that are associated with chemokine receptor expression or activity. Such compounds are provided herein.
……………………………………………………….

51 cas 1421063-65-1
succinate cas 1421065-31-7
52 cas 1416178-01-2
…………………………………..
http://www.google.com/patents/WO2013010453A1?cl=en
Example 74
1 -phenyl-4-[(2S,3aR,6aR)-3a- {[3-(trifluoromethyl)-7,8-dihydro- 1 ,6-naphthyridin-6(5H)- yl]carbonyl}octahydropentalen-2-yl]piperazin-2-one The title compound was obtained from the procedure of Example 63. Ή NMR
(400 MHz, CDCI3) δ ppm 8.70 (s, 1 H), 7.65 (s, 1 H), 7.35 (m, 2 H), 7.20 (m, 3 H), 4.60 – 4.90 (m, 2 H), 3.85 (m, 2H), 3.65 (m, 2H), 3.30 – 3.45 (m, 3H), 3.09 (m, 2H), 2.90 (m, 1H), 2.75 (m, 1H), 2.60 (m, 1H), 2.40 (m, 1H), 2.15 (m, 1H), 1.92 (m, 1H), 1.45 – 1.82 (m, 6H), 1.25 (m, 1H); MS (ESI) nVz 513 (M+H)+.

Example 75 C
l,5-anhydro-2,3-dideoxy-4-0-methyl-3-{[(2S,3aR,6aR)-3a-{[3-(trifluoromethyl)-7,8- dihydro- 1 ,6-naphthyridin-6(5H)-yl]carbonyl} octahydropentalen-2-yl]amino} -L-threo- pentitol
The title compound was prepared according to method A of Example 79G, substituting Example 75B for Example 79F, and isolated as the major isomer. Ή NMR (400 MHz, CDC13) δ 8.68 (s, 1H), 7.66 (s, 1H), 4.75 (m, 2H), 4.10 (m, 1H), 3.78 – 3.95 (m, 3H), 3.52 (m, 2H), 3.40 (s, 3H), 3.30 (m, 2H), 3.02 – 3.16 (m, 3H), 2.96 (m, 1H), 2.00 – 2.17 (m, 3H), 1.55 – 1.95 (m, 8H), 1.30 (m, 2H); MS (ESI) m/z 468 (M+H)+.

Example 76
l,5-anhydro-2,3-dideoxy-4-0-methyl-3-{[(2S,3aR,6aR)-3a-{[3-(trifluoromethyl)-7,8- dihydro- l,6-naphthyridin-6(5H)-yl]carbonyl}octahydropentalen-2-yl]amino}-D-erythro- pentitol
The title compound was prepared and purified according to the method A described in Example 79G, and was isolated as the major product. Ή NMR (400 MHz, CDC13) δ 8.70 (s, 1H), 7.70 (s, 1H), 4.70 – 4.90 (m, 2H), 4.10 (m, 1H), 3.80 – 3.95 (m, 3H), 3.35 – 3.50 (m, 2H), 3.42 (s, 3H), 3.30 (m, 2H), 3.15 (m, 2H), 3.05 (m, 1H), 2.75 (m, lH), 2.35 (m, lH), 2.15 (m, 1H), 1.50 – 2.00 (m, 9H), 0.90-1.10 (m, 2H). MS (ESI) m/z 468 (M+H)+.

Example 78
l,5-anhydro-2,3-dideoxy-4-0-methyl-3-{methyl[(2R,3aR,6aR)-3a-{[3-(trifluoromethyl)- 7,8-dihydro-l,6-naphthyridin-6(5H)-yl]carbonyl}octahydropentalen-2-yl]amino}-D- erythro-pentitol
To the solution of Example 79G (10 mg, 0.03 mmol) in dioxane (1 mL) was added formic acid (0.6 mL) and aqueous formalin solution (37%, 0.6 mL ). The mixture was heated at 80 °C overnight under nitrogen. The reaction mixture was concentrated in vaccum and was purified by HPLC to afford title compound as white solid (8.0 mg). Ή NMR (400 MHz, CD3OD) δ 8.70 (s, 1H), 8.06 (s, 1H), 4.75 – 4.90 (m, 2H), 4.30 (m, 1H), 3.90 – 4.10 (m, 4H), 3.50 – 3.72 (m, 4H), 3.45 (s, 3H), 3.15 (m, 2H), 2.78 (s, 3H), 2.60 (m, 1H), 1.85 – 2.20 (m, 9H), 1.80 (m, 1H), 1.58 (m, 1H), 1.42 (m, 1H); MS (ESI) m/z 482 (M+H)+.
To a solution of Example 78 (400 mg, 0.83 mmol) in MeOH (20 mL) was added succinic acid (98 mg 0.83 mmol) then the mixture was heated to 80 °C for 4 hours, then the solvent was concentrated under in vacuum to give the corresponding succinate salt (480 mg, 0.81 mmol, 96%). ‘H NMR (400 MHz, CD3OD) δ 8.70 (s, 1H), 8.05 (s, 1H), 4.85 (m, 2H), 4.25 (m, 1H), 4.05 (m, 2H), 3.95 (m, 2H), 3.70 (s, 1H), 3.50 (m, 2H), 3.42 (s, 3H), 3.33 (m, 1H), 3.12 (m, 2H), 2.70 (s, 3H), 2.55 (m, 1H), 2.50 (s, 4H), 2.15 (m, 3H), 1.70 – 2.02 (m, 7H), 1.55 (m, 1H), 1.40 (m, 1H).
ABS

4,4-dimethoxytetrahydro-2H-pyran
To the solution of dihydro-2H-pyran-4(3H)-one (50 g, 0.5 mol) in MeOH (500 mL) was added TiCl4 (1 g, 5 mmol) and Et3N (500 mg, 5 mmol), and the mixture stirred at room temperature for 10 hours, followed by the addition of Et3N (2 g, 20 mmol). The mixture was concentrated to 100 mL, diluted by methyl tert-butyl ether (1.5 L) and washed with ¾0 (500 mL), and brine. The organic layer was dried over Na2S04, filtered, and concentrated to yield Example 79A (60 g, 84% yield). Example 79B
4,4-dimethoxytetrahydro-2H-pyran compound with 4-methoxy-3,6-dihydro-2H-pyran To the solution of Example 79A (60 g, 0.42 mol) in dichloromethane (800 mL) at -78 °C was added TiCl4 (84 g, 0.42 mol). The mixture was stirred at -78 °C for 1 hour, followed by the addition of pyridine (66 g, 0.84 mol) and KOH (47 g, 0.84 mol). The mixture was stirred at -78 °C for additional 0.5 hour then warmed to room temperature and stirred overnight. The mixture was filtered. The filtrate was washed with water (1 L), dried over MgS04, filtered, and concentrated to yield Example 79B (52 g, 100%).
Example 79C
4,4-dimethoxytetrahydro-2H-pyran-3-ol
To the solution of Example 79B (2 g, 17.5 mmol) in MeOH (50 mL) was added meta-chloroperoxy benzoic acid (6 g, 35 mmol) in MeOH (6 mL) at 0 – 6 °C via addition funnel. After addition, the mixture was stirred at 0 °C for 4 hours. Upon reaction completion, the mixture was concentrated to yield white solid, which was then dissolved in dichloromethane (40 mL). Calcium hydroxide (14.8 g, 200 mmol) was added to the solution, and the solution was stirred for an additional 2 hours. The mixture was filtered and the filtrate was concentrated to yield crude title compound (2 g, 66.7%), which was used into next step without further purification.
Example 79D 3-methoxydihydro-2H-pyran-4(3H)-one
To the mixture of NaH (60%, 5.04 g, 12.6 mmol) in THF (200 mL) was added Example 79C (20 g, 12.6 mmol) in THF (150 mL). The mixture was stirred at 0 °C for 0.5 hour, followed by addition of iodomethane (200 g, 15.5 mmol), and was stirred overnight. HC1 (12 M, 12 mL) was added to the mixture, stirred at room temperature for additional 1.5 hours and concentrated. The residue was purified by column
chromatography to Example 79D (20 g, 100%).
Example 79E
(3S,4S)-3-methoxy-N-((S)-l-phenylethyl)tetrahydro-2H-pyran-4-amine To the solution of Example 79D (20 g, 153.8 mmol) and (S)- 1 -phenylethanamine
(18.6 g, 153.8 mmol) in dichloromethane (200 mL) was added Ti(i-OPr)4 (87.7 g, 310 mmol) and diisopropylethyl amine (40 g, 310 mmoL). The mixture was then stirred at room temperature for 18 hours, following by addition of sodium triacetoxyborohydride (65.1 g, 310 mmol), and MeOH (15 mL). The mixture was stirred for additional 4 hours, then poured into saturated NaHC(¾ solution, stirred for 1.5 hours and filtered. The filtrate was extracted with dichloromethane (2 x 200 mL) and concentrated. The residue was purified by preparative HPLC followed by chiral SFC separation to yield title compound (10 g, 27.7%).
Example 79F
(3S,4S)-3-methoxytetrahydro-2H-pyran-4-amine
To the solution of Example 79E (5.8 g, 21.3 mmol) in EtOH (100 mL) was added Pd/C (6 g), and the mixture was then submitted to hydrogenolysis at 50 °C (¾, 50 Psi) for 48 hours. The mixture was filtered and the filtrate was concentrated to yield 1.5 g (55%) of Example 79F which was used into next step directly without further purification.
Example 79G
l,5-anhydro-2,3-dideoxy-4-0-methyl-3- {[(2R,3aR,6aR)-3a-{[3-(trifluoromethyl)-7,8- dihydro- l,6-naphthyridin-6(5H)-yl]carbonyl}octahydropentalen-2-yl]amino}-D-erythro- pentitol
To the mixture of Example 1H (4.8 g, 13.6 mmol), Example 79F (1.5 g, 0.87 mmol) in dichloroethane (120 mL) was added Ti(i-OPr)4 (974 mg, 3.48 mmol), N,N- diisopropylethyl amine (1.2 g, 10 mmol). The mixture was then stirred overnight followed by addition of NaBH4 (132 mg, 3.48 mmol) and MeOH (5 mL). The mixture was stirred for another 12 hours before it was poured into saturated NaHC(¾, stirred at room temperature for 2 hours, filtered, and the filtrate was extracted by dichloromethane (3 x 200 mL). The organic layer was concentrated and the residue was purified by preparative HPLC to afford a mixture of two diastereomers. This mixture was purified by chiral SFC to yield Exmaple 76 as the first eluent, as well as the title compound (600 mg) as the second eluent. Ή NMR (400 MHz, CDC13) δ 8.70 (s, 1H), 7.70 (s, 1H), 4.68 – 4.85 (m, 2H), 4.22 (m, 1H), 4.05 (m, 1H), 3.70 – 3.90 (m, 3H), 3.55 (m, 2H), 3.35 – 3.45 (m, 2H), 3.40 (s, 3H), 3.30 (m, 1H), 3.10 (m, 2H), 2.45 (m, 1H), 2.30 (m, 1H), 2.00 – 2.20 (m, 3H), 1.55 – 1.95 (m, 6H), 1.30 (m, 2H); MS (ESI) m/z 468 (M+H)+.
The succinate salt of the above compound was prepared as follows: A mixture of free base of Example 79G (600 mg, 1.25 mmol), succinic acid (152 mg, 1.25 mmol) in EtOH (50 mL) was heated at 65 °C for 2 hours and then concentrated. The residue was washed with Et20 (25 mL) to yield white solid (610 mg, 86.4%). Ή NMR (400 MHz, CD3OD) δ 8.70 (s, 1H), 8.05 (s, 1H), 4.85 (m, 2H), 4.22 (d, J= 14.8 Hz, 1H), 3.95 (m, 3H), 3.70 (m, 1H), 3.30 – 3.53 (m, 4H), 3.38 (s, 3H), 3.12 (m, 2H), 2.55 (m, 1H), 2.52 (s, 4H), 2.15 (m, 1H), 1.70 – 2.05 (m, 9H), 1.55 (m, 1H), 1.38 (m, 1H).
Method B
Example 79H
( 1 R,4S)-methyl 4-aminocyclopent-2-enecarboxylate To a cooled mixture of (lR,4S)-2-azabicyclo[2.2.1]hept-5-en-3-one (13 g, 1 19 mmol) in MeOH (150 mL) was added SOCI2 (20 mL) dropwise to keep the temperature of the reaction under 15 °C. Upon completion of addition, the mixture was stirred at 5 °C for 3 hours. The solvent was removed under reduced pressure to yield liquid, which was dried under high vacuum to give Example 79H as white solid (23 g).
Example 791
(lR,4S)-methyl 4-(2,5 -dimethyl- lH-pyrrol-l-yl)cyclopent-2-enecarboxylate To a mixture of Example 79H (23 g, 163 mmol) in MeOH (100 ml) was added diisopropylethyl amine (23 g, 179 mmol) and acetyl acetone (20 g, 170 mmol), then the mixture was stirred at room temperature for 16 hours. The solvent was removed under reduced pressure, and the crude product was purified by column chromatography (S1O2, petroleum ethenEtOAc = 20: 1) to give Example 791 as yellow oil (20 g).
Example 79J (lR,4S)-methyl 1 -(3-bromopropyl)-4-(2,5-dimethyl- lH-pyrrol- 1 -yl)cyclopent-2- enecarboxylate
To a solution of Example 791 (16.5 g, 74.4 mmol) in THF (200 ml) was added dropwise lithium hexamethyl bis(trimethylsilyl)amide (1 M, 1 19 mL) at -50 °C, stirredd for 1 hour, allowed to warm to -20 °C, followed by the dropwise addition of 1,3- dibromopropane (150 g, 744 mmol) over 1 hour. The reaction mixture was stirred at -20 °C for 1 hour, quenched with aqueous NH4C1 solution (6%, 600 mL), and extracted with ethyl acetate. The organic layer was washed with NH4CI solution, brine, dried over Na2S04, filtered, and concentrated. The residued was purified by silica gel column chromatography (petroleum ethenEtOAc = 80: 1) to give Example 79J (16 g).
Example 79K
(2R,3aR,6aR)-methyl 2-(2,5-dimethyl- lH-pyrrol- 1 -yl)octahydropentalene-3a-carboxylate To a solution of compound 79 J (16 g, 47 mmol) and azobisisobutyronitrile (1.6 g, 10 mmol) in toluene (1.8 L) was added a solution of tributyl tin hydride (32 mL) in toluene (200 mL) at 1 10 °C over 1 hour. After refluxing for 3 hours, the reaction mixture was quenched by saturated aqueous KF (200 mL), and extracted with ethyl acetate. The organic layer was washed with brine, dried over Na2S04;filtered, and concentrated. The residue was purified by column chromatography (S1O2, petroleum ethenEtOAc = 50: 1) to give compound Example 79K (8 g) as white solid.
Example 79L
(2R,3aR,6aR)-2-(2,5-dimethyl-lH-pyrrol-l-yl)octahydropentalene-3a-carboxylic acid To a solution of Example 79K (5.3 g, 20.3 mmol) in MeOH (33 mL) and water (15 mL) was added a aqueous solution of NaOH (3.2 g, 80 mmol in 4 mL water) and the mixture was heated at 65 °C for 16 hours. The mixture was cooled to room temperature, adjusted the pH to about 4 with HC1 solution (4 N) and filtered to collect Example 79L (4.5 g) as yellow solid and used in next step without purification.
Example 79M
((2R,3aR,6aR)-2-(2,5-dimethyl- lH-pyrrol- 1 -yl)octahydropentalen-3a-yl)(3- (trifluoromethyl)-7,8-dihydro-l,6-naphthyridin-6(5H)-yl)methanone To a solution of compound Example 79L (5 g, 20.2 mmol) in dichloromethane (50 mL) was added hydroxybenzotriazole (4.2 g, 30.9 mmol), l-ethyl-3-(3- dimethylaminopropyl) carbodiimide hydrochloride (5.8 g, 30.4 mmol), and Et3N (6.0 g, 59.4 mmol), and the mixture was stirred at room temperature for 16 hours. The reaction mixture was suspended in water and extracted with dichloromethane (3 x 300 mL). The combined organic layer was washed with brine, dried over Na2S04, filtered, and concentrated to result in the title compound (8 g), which was used in the next step without purification.
Example 79N
((2R,3aR,6aR)-2-aminooctahydropentalen-3a-yl)(3-(trifluoromethyl)-7,8-dihydro- l,6- naphthyridin-6(5H)-yl)methanone
To a solution of Example 79M (8.0 g, 18.6 mmol) in MeOH (100 mL) was added hydroxylamine hydrochloride (8.0 g, 1 15.9 mmol), 50% hydroxylamine hydrate (12 mL) and ¾0 (40 mL). The mixture was heated at reflux for 13 hours and cooled to room temperature. The mixture was treated with NaOH (10 N) to adjust the pH to about 1 1, and extracted with dichloromethane (3 x 300 mL). The combined organic layer was washed with brine, dried over MgS04, filtered, and concentrated. A solution of HCl in EtOAc (30 mL) was added to the residue with stirring at room temperature for 1 hour. The solvent was removed and the HCl salt of Example 79N (6.5 g) was used for next step without purification.
Example 790
tert-butyl(3,6-dihydro-2H-pyran-4-yloxy)dimethylsilane To a mixture of the tetrahydro-4H-pyran-4-one (38.9 g, 0.38 mol) and Et3N (76.8 g, 0.76 mol) in dichloromethane (800 mL) was added trimethyl trifluoromethanesulfonate (105.5 g, 0.399 mol) dropwise over 3 hours. After addition, the reaction was allowed to warm to room temperature and stirred overnight. Water was added and the resulting solution was extracted with dichloromethane (2 x 500 mL). The combined organic phase was washed with water (2 x 500 mL) and brine (2 x 200 mL), dried over Na2S04, filtered, and concentrated to give Example 790 (78 g, 85%) as an oil.
Example 79P
sodium (3R)-3,4-dihydroxytetrahydro-2H-pyran-4-sulfonate To a solution of (DHQD^PHAL (hydroquinidein 1 ,4-phthalazinediyl diether) (3.06 g, 3.93 mol), K20s04 (723 mg, 1.96 mol) and N-methylmorpholine-N-oxide (58.4 g, 0.432 mol) in acetone/H20 (700 mL, 10/1) at 0 °C was added slowly a solution of Example 790 (84 g, 0.393 mol) in acetone (100 mL) for 5 hours. The resulting solution was stirred at 10-20 °C overnight. A freshly prepared solution of Na2S20s (44.8 g, 0.236 mol) in water (315 mL) was added followed by acetic acid (67.3 mL). After stirring for 16 hours at room temperature, the solid was filtered and washed with isopropanol (400 mL) and dried to provide Example 79P (60 g, 73%) as a white solid.
Example 79Q
(R)-4,4-dimethoxytetrahydro-2H-pyran-3-ol To a solution of Example 79P (60 g, 0.294 mol) and HC(OCH3)3 (69.3 g, 0.647 mol) in MeOH (500 mL) at 50 °C was added HCl/MeOH (68 mL, 5-6 N) slowly over 30 minutes. Then the slurry was cooled to 5 °C and 50% of NaOH (100 mL in water) was added over 1 hour. The solid was filtered and the filtrate was concentrated. The resulting solution was washed with toluene for several times and then concentrated to give Example 79Q (38 g, yield: 88%) as an oil.
Example 79R
(R)-3-methoxydihydro-2H-pyran-4(3H)-one To a solution of Example 79Q (9.5 g, 64.68 mol) in THF (300 mL) was added sodium tert-butoxide (9.3 g, 97.02 mmol) at ice bath. Then dimethyl sulfate (13.4 g, 106 mmol) was added over 20 minutes, maintaining an interal temp below 36 °C. After addition, the reaction mixture was stirred for 4 hours at room temperature. Water (200 mL) was added followed by addition of 2N HC1 (100 mL). The apparent pH is below 1. After 16 hours of reaction, NaHC(¾ (20 g) was added and the mixture was extracted with EtOAc (4 x 300 mL), dried over Na2S04, filtered, and concentrated to give Example 79R (6 g, yield: 71 %) as an oil.
Example 79G
l,5-anhydro-2,3-dideoxy-4-0-methyl-3- {[(2R,3aR,6aR)-3a-{[3-(trifluoromethyl)-7,8- dihydro- l,6-naphthyridin-6(5H)-yl]carbonyl}octahydropentalen-2-yl]amino}-D-erythro- pentitol
A solution of Example 79N (3.2 g, 9.0 mmol) in isopropyl acetate (80 mL) was cooled with ice bath and tributylamine (3.3 g, 20.7 mmol) was added dropwise, followed by the addition of isopropyl alcohol (1.6 ml, 20.7 mmol). Sodium triacetoxyborohydride (4.4 g, 20.7 mmol) was added. After 1 hour at room temperature, a solution of Exampole 79R (1.75 g, 13 mmol) in isopropyl acetate (10 mL) was added to the mixture at 1 °C. Then the mixture was stirred at room temperature for 15 hours and partitioned between satureated aqueous NaHC(¾ (80 mL), water (50 mL) and EtOAc (400 mL). The aqueous phase was further extracted with EtOAc (200 mL). The combined organic phase was washed with saturated.aqeous NaHC03 solution, dried with Na2S04, filtered, concentrated. The residue was purified by column chromatography (S1O2,
dichloromethane:MeOH = 20: 1) to give Example 79G as a mixture of diastereomers, which was then further purified by chiral SFC to yield title compound as the first eluent and white solid upon concentration, as well as Example 80 as the second eluent.

…………………………………………………………
Bioorganic & Medicinal Chemistry Letters, 23(1), 351-354; 2013
http://www.sciencedirect.com/science/article/pii/S0960894X12013601

Regents and conditions: (a) (Boc)2O, DMAP, THF, 58 °C; (b) AcOCH2C(
……………………………
http://pubs.acs.org/doi/full/10.1021/op500265z

………………………………..

YM758 Monophosphate, A Novel If Channel Inhibitor


| N-[2-[(3R)-3-[(3,4-Dihydro-6,7-dimethoxy-2(1H)-isoquinolinyl)carbonyl]-1-piperidinyl]ethyl]-4-fluorobenzamide Phosphate; (R)-(-)-N-[2-[3-[(6,7-Dimethoxy-1,2,3,4 -tetrahydroisoquinolin-2-yl)carbonyl]piperidino]ethyl]-4-fluorobenzamide monophosphate; YM 758; |
| CAS Number: 312752-86-6 |
U.S. Patent No. 6,573,279, incorporated herein by reference, describes isoquinoline compounds with 1 channel blocker activity and their use in treating a variety of cardiovascular diseases. U.S. Patent Application Publication Nos. 20060084807 and
20070129357, each of which is incorporated herein by reference, describe methods for making those isoquinoline compounds as well as crystals of certain fluorobenzamide derivatives of them. U.S. Patent Publication No. 20090247572, incorporated herein by reference, relates to the use of one of these isoquinoline fluorobenzamide derivatives, (-)-N- {2-[(i?)-3-(6,7-dimethoxy-l ,2,3,4-tetrahydroisoquinoline-2-carbonyl)piperidino]ethyl}-4- fluorobenzamide monophosphate (referred to in that patent publication as “compound A” and “chemical formulation I” and referred to herein as “YM758”), for treating atrial fibrillation.
To date, however, these isoquinoline compounds in general and YM758 in particular have not been developed as cardiovascular drugs. Thus, there remains a need for methods of using these compounds, alone and in combination with other cardiovascular drugs, to treat cardiovascular disease, as well as a need for pharmaceutical formulations and unit dose forms useful in such methods. This invention meets those needs.
Provided herein are fast-acting (immediate release) and modified (sustained) release oral formulations as well as intravenous formulations of YM758. The present invention also provides unit dose forms of these formulations. The present invention also provides methods for using these formulations and unit dose forms alone and in combination with other drugs for the treatment of cardiovascular disease, including but not limited to stable angina, atrial fibrillation, and heart failure. In these methods, the pharmaceutical formulations and unit dose forms of the invention may be dosed alone or in combination with other drugs, including but not limited to drugs such as beta-blockers, anti-arrhythmia drugs, calcium channel blockers, sodium channel blockers, potassium channel blockers, adenosine, and digitalis. The invention also provides formulations and unit dose forms of YM758 and another drug selected from the group of drugs including beta-blockers, anti-arrhythmia drugs, calcium channel blockers, sodium channel blockers, potassium channel blockers, adenosine, and digitalis. The single agent and combination pharmaceutical formulations and unit dose forms of the invention include capsule, tablet, and solution formulations and unit dose forms that provide either immediate or sustained release. The pharmaceutical formulations in solution forms are, in various embodiments, suitable for intravenous, subcutaneous, intraperitoneal, and intramuscular administration.
Thus, in one aspect, the present invention provides an oral formulation comprising or consisting essentially of YM758 and optionally an excipient. As used herein, the excipient is suitable for administration to human patients with various cardiovascular diseases and includes, without limitation, one or more of the following: an additive, an anti- foaming agent, a binder, a chemical stabilizer, a coloring agent, a diluent, a disintegrating agent, an emulsifying agent, a filler, a flavoring agent, a glidant, a lubricant, a pH modifier, a plasticizer, a solubilizer, a swelling enhancer, a spheronization aid, a solubility enhancer, and a suspending agent. In some embodiments, the formulation is provided in a unit dose form, which may be, for example, a tablet or capsule. In various embodiments, the unit dose forms contain from about 5 mg to about 80 mg of YM758. In some embodiments, the unit dose forms contain from about 5 mg to about 50 mg of YM758. In other embodiments, the unit dose form contains from about 10 mg to about 40 mg of YM758. In one embodiment, the unit dose form contains about 25 mg of YM758.
In another aspect, the present invention provides formulations comprising or consisting essentially of YM758 and optionally an excipient that are suitable for intravenous, subcutaneous, intraperitoneal, and intramuscular administration. As used herein, the excipient is suitable for administration to human cardiovascular disease patients and includes, without limitation, one or more of the following: an additive, an anti-foaming agent, a chemical stabilizer, a diluent, an emulsifying agent, a pH modifier, a buffering agent, an osmolality modifier, a salt, a solubilizer, a solubility enhancer, and a suspending agent. In some embodiments, the formulation is a solution formulation. In one embodiment, the solution formulation is provided in a unit dose form, which may be, for example, in a vial, an ampoule or an intravenous bag. In various embodiments, the unit dose forms contain from about 5 mg to about 80 mg of YM758. In some embodiments, the unit dose forms contain from about 5 mg to about 50 mg of YM758. In another embodiment, the unit dose form contains from about 10 mg to about 40 mg of YM758. In one embodiment, the unit dose form contains about 25 mg of YM758. [0008] In various embodiments, the oral formulation is an immediate release formulation, including, and unit dose forms of this formulation include, without limitation, a gelatin capsule comprising a YM758 formulation.
NMR: δ 1.25-1.50 (1H, m), 1.53-1.76 (3H, m), 2.15-2.80 (6H, m), 2.95-3.10 (3H m), 3.40-3.50 (2H, m), 3.60-3.75 (8H, m), 4.50 (1H, q), 4.63 (1H, q), 6.73 (1H, s), 6.78, 6.85 (1H, s in combination), 7.29 (2H, t), 7.91-7.95 (2H, m), 8.60 (1H, br).
FAB-MS m/z: 470 (M++1).


A novel, practical, and efficient synthesis of (−)-N-{2-[(R)-3-(6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline-2-carbonyl)piperidino]ethyl}-4-fluorobenzamide monophosphate (YM758 monophosphate, (R)-1·H3PO4 (Figure 1) is described. The target molecule (R)-1 has a potent If current channel inhibitor. Medicinal chemistry synthetic routes were very long and suffered from extensive use of chlorinated solvents and silica-gel column chromatography. A number of steps in the medicinal chemistry route were also unattractive for large-scale synthesis due to some reasons for example the use of unstable intermediates.
An important objective of a new synthetic route was avoidance of such a use of unstable intermediate, and it was achieved by the discovery of an important 4,5-dihydrooxazole intermediate 19 and ring-opening N-alkylation of chiral amine with 19 under acidic condition. The new procedure does not require any purification by column chromatography for all steps.
The overall yield was significantly improved from 14% or 34% to 49% compared to that of the medicinal synthetic routes. This highly efficient process was successfully demonstrated at a pilot-scale operation, yielding 36.5 kg of (R)-1·H3PO4.
(−)-N-{2-[(R)-3-(6,7-Dimethoxy-1,2,3,4-tetrahydroisoquinoline-2-carbonyl)piperidino]ethyl}-4-fluorobenzamide Monophosphate (YM758 Monophosphate, (R)-1·H3PO4)
Cefditoren pivoxil


Cefditoren pivoxil
ME-1207, Spectracef, Meiact
117467-28-4
- (-)-(6R,7R)-2,2-dimethylpropionyloxymethyl 7-((Z)-2-(2-aminothiazol-4-yl)-2-methoxyiminoacetamido)-3-((Z)-2-(4-methylthiazol-5-yl)ethenyl)-8-oxo-5-thia-1-azabicyclo(4.2.0)oct-2-ene-2-carboxylate
- 7-(2-(2-aminothiazol-4-yl)-2-(methoxyimino)acetamido)-3-(2-(4-methylthiazol-5-yl)ethenyl)cephem-4-carboxylic acid pivaloyloxymethyl ester
- CDTR-PI
- cefditoren pivoxil
- ME 1207
- ME-1207
- Spectracef
Novel crystalline form of cefditoren pivoxil (first disclosed in EP175610). Represents Yungjin Pharma’s first interest in this API, which was developed and launched by Meiji Seika and previous licensee TAP Pharmaceuticals, and now marketed by Merus Labs, for treating chronic bronchitis and community acquired pneumonia caused by bacterial infections
Cefditoren is a third-generation cephalosporin antibiotic for oral use. It is commonly marketed under the trade name Spectracef by Vansen Pharma Inc.
Cefditoren is also marketed under the name Meiact by Meiji Seika Pharma Co., Ltd.[1]
Spectrum of bacterial susceptibility
Cefditoren has a broad spectrum of activity and has been used to treat bacterial infections of the skin and respiratory tract, including bronchitis, pneumonia, and tonsillitis. The following represents MIC susceptibility data for a few medically significant microorganisms.
- Haemophilus influenzae: ≥0.063 – 0.25 μg/ml
- Staphylcoccus aureus: 0.25 – >128 μg/ml (includes MRSA)
- Streptococcus pyogenes: ≤0.004 – 2 μg/ml[2]
Cefditoren is a broad-spectrum antibiotic against Gram-negative and Gram-positive bacteria, but does not have antibacterial activity against Pseudomonas aeruginosa.[3]
Clinical use
Indications
Cefditoren is used to treat uncomplicated skin and skin structure infections, community-acquired pneumonia, acute bacterial exacerbation of chronic bronchitis, pharyngitis, and tonsillitis.
Formulations
Cefditoren is available as 200- and 400-mg tablets. It can be formulated as the prodrug cefditoren pivoxil.

Chemical structure of cefditoren pivoxil

References
- Meiact Full Description
- http://www.toku-e.com/Assets/MIC/Cefditoren%20sodium.pdf
- “Disease relevance of Cefditoren”. Retrieved June 24, 2014.
- Chem Pharm Bull 1992,39(9),2433
- J Antibiot 1990,43(8),1047
Synthesis Reference
Kiyoshi Yasui, Masahiro Onodera, Masamichi Sukegawa, Tatsuo Watanabe, Yuichi Yamamoto, Yasushi Murai, Katsuharu Iinuma, “Crystalline substance of cefditoren pivoxyl and the production of the same.” U.S. Patent US6294669, issued March, 1986.
| Patent | Submitted | Granted |
|---|---|---|
| Therapy for Treating Resistant Bacterial Infections [US2009275552] | 2009-11-05 | |
| Process for the preparation of thiazole intermediate [US6833459] | 2003-10-30 | 2004-12-21 |
| Nanoparticulate and Controlled Release Compositions Comprising Cefditoren [US8119163] | 2008-11-13 | 2012-02-21 |
External links
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (7R)-7-((Z)-2-(2-Aminothiazol-4-yl)-2-(methoxyimino)acetamido)-3-((Z)-2-(4-methylthiazol-5-yl)vinyl)-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid | |
| Clinical data | |
| Trade names | Spectracef |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a605003 |
| Legal status |
?
|
| Identifiers | |
| CAS number | 104145-95-1 |
| ATC code | J01DD16 |
| PubChem | CID 9870843 |
| DrugBank | DB01066 |
| ChemSpider | 8046534 |
| UNII | 81QS09V3YW |
| Chemical data | |
| Formula | C19H18N6O5S3 |
| Mol. mass | 506.58 g/mol |
WO-2014189308, Yungjin Pharmaceutical Co Ltd
