PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
A long-acting decanoateester of haloperidol is used as an injection given every four weeks to people with schizophrenia or related illnesses who have poor adherence to medication regimens (most commonly due to them forgetting to take their medication, or due to poor insight into their illness) and suffer frequent relapses of illness, or to overcome the drawbacks inherent to its orally administered counterpart.[6] Such long acting injections are controversial because it can be seen as denying people their right to stop taking their medication.
Haloperidol was discovered by Paul Janssen.[70] It was developed in 1958 at the Belgian company Janssen Pharmaceutica and submitted to the first of clinical trials in Belgiumlater that year.[71]
Coincident with civil unrest in the United States in the 1960s and 1970s, schizophrenia was racialized to match the behavior of angry/violent black men. Haldol was promoted as a way to pacify them, and was marketed to appeal to feelings of racial unease. (cf. Metzl 2010. The Protest Psychosis)
Soviet dissidents, including medical staff, have reported several times on the use of haloperidol in the Soviet Union for punitive purposes or simply to break the prisoners’ will.[72][73][74] Notable dissidents who were administered haloperidol as part of their court-ordered treatment were Sergei Kovalev and Leonid Plyushch.[75] The accounts Plyushch gave in the West, after he was allowed to leave the Soviet Union in 1976, were instrumental in triggering Western condemnation of Soviet practices at the World Psychiatric Association‘s 1977 meeting.[76] The use of haloperidol in the Soviet Union’s psychiatric system was prevalent because it was one of the few psychotropic drugs produced in quantity in the USSR.[77]
Haloperidol has been used for its sedating effects during the deportations of immigrants by the United States Immigration and Customs Enforcement (ICE). During 2002-2008, federal immigration personnel used haloperidol to sedate 356 deportees. By 2008, following court challenges over the practice, it was given to only three detainees. Following lawsuits, U.S. officials changed the procedure so the drug is administered only by the recommendation of medical personnel and under court order.[78][79]
Brand names
Haloperidol is sold under the tradenames Aloperidin, Bioperidolo, Brotopon, Dozic, Duraperidol (Germany), Einalon S, Eukystol, Haldol (common tradename in the US and UK), Halosten, Keselan, Linton, Peluces, Serenace and Sigaperidol.
Veterinary use
Haloperidol is also used on many different kinds of animals. It appears to be particularly successful when given to birds, e.g., a parrot that will otherwise continuously pluck its feathers out.[80]
Jump up^ Joint Formulary Committee (2013). British National Formulary (BNF) (65 ed.). London, UK: Pharmaceutical Press. p. 229-230. ISBN978-0-85711-084-8. edit
^ Jump up to:ab Brayfield, A, ed. (13 December 2013). “Haloperidol”. Martindale: The Complete Drug Reference. London, UK: Pharmaceutical Press. Retrieved 29 May 2014.
Jump up^ Rossi, S, ed. (2013). Australian Medicines Handbook (2013 ed.). Adelaide: The Australian Medicines Handbook Unit Trust. ISBN978-0-9805790-9-3. edit
Jump up^ Giannini, A. James; Underwood, Ned A.; Condon, Maggie (2000). “Acute Ketamine Intoxication Treated by Haloperidol”. American Journal of Therapeutics7 (6): 389–91.doi:10.1097/00045391-200007060-00008. PMID11304647.
Jump up^ Giannini, A. James; Eighan, Michael S.; Loiselle, Robert H.; Giannini, Matthew C. (1984). “Comparison of Haloperidol and Chlorpromazine in the Treatment of Phencyclidine Psychosis”. The Journal of Clinical Pharmacology24 (4): 202–4.doi:10.1002/j.1552-4604.1984.tb01831.x. PMID6725621.
Jump up^ Cavanaugh, SV (1986). “Psychiatric emergencies”. The Medical clinics of North America70 (5): 1185–202. PMID3736271.
Jump up^ Irving, Claire B; Adams, Clive E; Lawrie, Stephen (2006). “Haloperidol versus placebo for schizophrenia”. In Irving, Claire B. Cochrane Database of Systematic Reviews (4): CD003082. doi:10.1002/14651858.CD003082.pub2. PMID17054159.
Jump up^ Allen, MH; Currier, GW; Hughes, DH; Reyes-Harde, M; Docherty, JP; Expert Consensus Panel for Behavioral Emergencies (2001). “The Expert Consensus Guideline Series. Treatment of behavioral emergencies”. Postgraduate Medicine (Spec No): 1–88; quiz 89–90. PMID11500996.
Jump up^ Allen, Michael H.; Currier, Glenn W.; Hughes, Douglas H.; Docherty, John P.; Carpenter, Daniel; Ross, Ruth (2003). “Treatment of Behavioral Emergencies: A Summary of the Expert Consensus Guidelines”. Journal of Psychiatric Practice9 (1): 16–38. doi:10.1097/00131746-200301000-00004. PMID15985913.
Jump up^ Allen, Michael H.; Currier, Glenn W.; Carpenter, Daniel; Ross, Ruth W.; Docherty, John P. (2005). “Introduction: Methods, Commentary, and Summary”. Journal of Psychiatric Practice11: 5. doi:10.1097/00131746-200511001-00002.
Jump up^ Oosthuizen, P.; Emsley, R. A.; Turner, J.; Keyter, N. (2001). “Determining the optimal dose of haloperidol in first-episode psychosis”. Journal of Psychopharmacology15 (4): 251–5. doi:10.1177/026988110101500403. PMID11769818.
Smith, Thomas J.; Ritter, Joseph K.; Poklis, Justin L.; Fletcher, Devon; Coyne, Patrick J.; Dodson, Patricia; Parker, Gwendolyn (2012). “ABH Gel is Not Absorbed from the Skin of Normal Volunteers”. Journal of Pain and Symptom Management43(5): 961–6. doi:10.1016/j.jpainsymman.2011.05.017. PMID22560361.
Weschules, Douglas J. (2005). “Tolerability of the Compound ABHR in Hospice Patients”. Journal of Palliative Medicine8 (6): 1135–43.doi:10.1089/jpm.2005.8.1135. PMID16351526.
Jump up^ Truven Health Analytics, Inc. DrugPoint® System (Internet) [cited 2013 Sep 29]. Greenwood Village, CO: Thomsen Healthcare; 2013.
Jump up^ Joint Formulary Committee. British National Formulary (BNF) 65. Pharmaceutical Pr; 2013.
^ Jump up to:ab Leucht, Stefan; Cipriani, Andrea; Spineli, Loukia; Mavridis, Dimitris; Örey, Deniz; Richter, Franziska; Samara, Myrto; Barbui, Corrado; Engel, Rolf R; Geddes, John R; Kissling, Werner; Stapf, Marko Paul; Lässig, Bettina; Salanti, Georgia; Davis, John M (2013). “Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: A multiple-treatments meta-analysis”. The Lancet382 (9896): 951–62.doi:10.1016/S0140-6736(13)60733-3. PMID23810019.
^ Jump up to:ab Silvestri, Simone; Seeman, Mary V.; Negrete, Juan-Carlos; Houle, Sylvain; Shammi, C.M.; Remington, Garry J.; Kapur, Shitij; Zipursky, Robert B.; Wilson, Alan A.; Christensen, Bruce K.; Seeman, Philip (2000). “Increased dopamine D 2 receptor binding after long-term treatment with antipsychotics in humans: A clinical PET study”.Psychopharmacology152 (2): 174–80. doi:10.1007/s002130000532.PMID11057521.
Jump up^ Dorph-Petersen, Karl-Anton; Pierri, Joseph N; Perel, James M; Sun, Zhuoxin; Sampson, Allan R; Lewis, David A (2005). “The Influence of Chronic Exposure to Antipsychotic Medications on Brain Size before and after Tissue Fixation: A Comparison of Haloperidol and Olanzapine in Macaque Monkeys”. Neuropsychopharmacology30 (9): 1649–61. doi:10.1038/sj.npp.1300710. PMID15756305.
Jump up^ Vernon, Anthony C.; Natesan, Sridhar; Modo, Mike; Kapur, Shitij (2011). “Effect of Chronic Antipsychotic Treatment on Brain Structure: A Serial Magnetic Resonance Imaging Study with Ex Vivo and Postmortem Confirmation”. Biological Psychiatry69 (10): 936–44. doi:10.1016/j.biopsych.2010.11.010. PMID21195390.
Jump up^ Dalton, Susanne Oksbjerg; Mellemkjaer, Lene; Thomassen, Lars; Mortensen, Preben B.; Johansen, Christoffer (2005). “Risk for cancer in a cohort of patients hospitalized for schizophrenia in Denmark, 1969–1993″. Schizophrenia Research75 (2–3): 315–24.doi:10.1016/j.schres.2004.11.009. PMID15885523.
Jump up^ Grinshpoon, Alexander; Barchana, Micha; Ponizovsky, Alexander; Lipshitz, Irena; Nahon, Daniella; Tal, Orna; Weizman, Abraham; Levav, Itzhak (2005). “Cancer in schizophrenia: Is the risk higher or lower?”. Schizophrenia Research73 (2–3): 333–41.doi:10.1016/j.schres.2004.06.016. PMID15653279.
Jump up^ Szarfman, Ana; Tonning, Joseph M; Levine, Jonathan G; Doraiswamy, P. Murali (2006). “Atypical Antipsychotics and Pituitary Tumors: A Pharmacovigilance Study”.Pharmacotherapy26 (6): 748–58. doi:10.1592/phco.26.6.748. PMID16716128.
Jump up^ Hippisley-Cox, Julia; Vinogradova, Y; Coupland, C; Parker, C (2007). “Risk of Malignancy in Patients with Schizophrenia or Bipolar Disorder”. Archives of General Psychiatry64 (12): 1368–76. doi:10.1001/archpsyc.64.12.1368. PMID18056544.
Jump up^ Levav, Itzhak; Kohn, Robert; Barchana, Micha; Lipshitz, Irena; Pugachova, Inna; Weizman, Abraham; Grinshpoon, Alexander (2009). “The risk for cancer among patients with schizoaffective disorders”. Journal of Affective Disorders114 (1–3): 316–20.doi:10.1016/j.jad.2008.06.010. PMID18675461.
Jump up^ Sandyk, R; Hurwitz, MD (1983). “Toxic irreversible encephalopathy induced by lithium carbonate and haloperidol. A report of 2 cases”. South African medical journal64 (22): 875–6. PMID6415823.
Jump up^ Bush, S. E.; Hatton, R. C.; Winterstein, A. G.; Thomson, M. R.; Woo, G. W. (2008). “Effects of concomitant amiodarone and haloperidol on Q-Tc interval prolongation”.American Journal of Health-System Pharmacy65 (23): 2232–6.doi:10.2146/ajhp080039. PMID19020191.
Jump up^ Igarashi, K.; Kasuya, F.; Fukui, M.; Usuki, E.; Castagnoli Jr, N. (1995). “Studies on the metabolism of haloperidol (HP): The role of CYP3A in the production of the neurotoxic pyridinium metabolite HPP+ found in rat brain following ip administration of HP”. Life Sciences57 (26): 2439–46. doi:10.1016/0024-3205(95)02240-5. PMID8847965.
Jump up^ Usuki, Etsuko; Pearce, Robin; Parkinson, Andrew; Castagnoli, Neal (1996). “Studies on the Conversion of Haloperidol and Its Tetrahydropyridine Dehydration Product to Potentially Neurotoxic Pyridinium Metabolites by Human Liver Microsomes”. Chemical Research in Toxicology9 (4): 800–6. doi:10.1021/tx960001y. PMID8831826.
Jump up^ Avent, Kathryn M.; Devoss, J. J.; Gillam, Elizabeth M. J. (2006). “Cytochrome P450-Mediated Metabolism of Haloperidol and Reduced Haloperidol to Pyridinium Metabolites”.Chemical Research in Toxicology19 (7): 914–20. doi:10.1021/tx0600090.PMID16841959.
Jump up^ Avent, Kathryn M.; Riker, Richard R.; Fraser, Gilles L.; Van Der Schyf, Cornelis J.; Usuki, Etsuko; Pond, Susan M. (1997). “Metabolism of haloperidol to pyridinium species in patients receiving high doses intravenously: Is HPTP an intermediate?”. Life Sciences61 (24): 2383–90. doi:10.1016/S0024-3205(97)00955-7. PMID9399630.
Jump up^ Kawashima, Hidekazu; Iida, Yasuhiko; Kitamura, Youji; Saji, Hideo (2004). “Binding of 4-(4-chlorophenyl)-1-[4-(4-fluorophenyl)-4-oxobutyl]pyridinium ion (HPP+), a metabolite of haloperidol, to synthetic melanin: Implications for the dopaminergic neurotoxicity of HPP+“. Neurotoxicity Research6 (7–8): 535–42. doi:10.1007/BF03033449.PMID15639785.
Jump up^ Bishnoi, Mahendra; Chopra, Kanwaljit; Kulkarni, Shrinivas K. (2008). “Activation of striatal inflammatory mediators and caspase-3 is central to haloperidol-induced orofacial dyskinesia”. European Journal of Pharmacology590 (1–3): 241–5.doi:10.1016/j.ejphar.2008.06.033. PMID18590723.
Jump up^ Eyles, Darryl W.; Avent, Kathryn M.; Stedman, Terry J.; Pond, Susan M. (1997). “Two pyridinium metabolites of haloperidol are present in the brain of patients at post-mortem”.Life Sciences60 (8): 529–34. doi:10.1016/S0024-3205(96)00656-X. PMID9042387.
Jump up^ Ulrich, Sven; Neuhof, Sabine; Braun, Verena; Danos, Peter; Pester, Uwe; Hoy, Ludwig (2000). “Disposition of Haloperidol Pyridinium and Reduced Haloperidol Pyridinium in Schizophrenic Patients: No Relationship with Clinical Variables During Short-Term Treatment”. Journal of Clinical Psychopharmacology20 (2): 210–9.doi:10.1097/00004714-200004000-00014. PMID10770460.
Jump up^ Ulrich, S.; Sandmann, U.; Genz, A. (2005). “Serum Concentrations of Haloperidol Pyridinium Metabolites and the Relationship with Tardive Dyskinesia and Parkinsonism: A Cross-Section Study in Psychiatric Patients”. Pharmacopsychiatry38 (4): 171–7.doi:10.1055/s-2005-871240. PMID16025420.
Jump up^ Seeman, P; Tallerico, T (1998). “Antipsychotic drugs which elicit little or no Parkinsonism bind more loosely than dopamine to brain D2 receptors, yet occupy high levels of these receptors”. Molecular Psychiatry3 (2): 123–34.doi:10.1038/sj.mp.4000336. PMID9577836.
Jump up^ Leysen, JE; Janssen, PM; Megens, AA; Schotte, A (1994). “Risperidone: A novel antipsychotic with balanced serotonin-dopamine antagonism, receptor occupancy profile, and pharmacologic activity”. The Journal of Clinical Psychiatry55 (Suppl): 5–12.PMID7520908.
Jump up^ Cobos, Enrique J.; Del Pozo, Esperanza; Baeyens, José M. (2007). “Irreversible blockade of sigma-1 receptors by haloperidol and its metabolites in guinea pig brain and SH-SY5Y human neuroblastoma cells”. Journal of Neurochemistry102 (3): 812–25.doi:10.1111/j.1471-4159.2007.04533.x. PMID17419803.
Jump up^ Colabufo, Nicolaantonio; Berardi, Francesco; Contino, Marialessandra; Niso, Mauro; Abate, Carmen; Perrone, Roberto; Tortorella, Vincenzo (2004). “Antiproliferative and cytotoxic effects of some σ2 agonists and σ1 antagonists in tumour cell lines”. Naunyn-Schmiedeberg’s Archives of Pharmacology370 (2): 106–13. doi:10.1007/s00210-004-0961-2. PMID15322732.
^ Jump up to:abcdefghijk Kroeze, Wesley K; Hufeisen, Sandra J; Popadak, Beth A; Renock, Sean M; Steinberg, Seanna; Ernsberger, Paul; Jayathilake, Karu; Meltzer, Herbert Y; Roth, Bryan L (2003). “H1-Histamine Receptor Affinity Predicts Short-Term Weight Gain for Typical and Atypical Antipsychotic Drugs”. Neuropsychopharmacology28 (3): 519–26.doi:10.1038/sj.npp.1300027. PMID12629531.
^ Jump up to:ab Kornhuber, Johannes; Schultz, Andreas; Wiltfang, Jens; Meineke, Ingolf; Gleiter, Christoph H.; Zöchling, Robert; Boissl, Karl-Werner; Leblhuber, Friedrich; Riederer, Peter (1999). “Persistence of Haloperidol in Human Brain Tissue”. The American Journal of Psychiatry156 (6): 885–90. PMID10360127.
Jump up^ Kornhuber, Johannes; Wiltfang, Jens; Riederer, Peter; Bleich, Stefan (2006). “Neuroleptic drugs in the human brain: Clinical impact of persistence and region-specific distribution”. European Archives of Psychiatry and Clinical Neuroscience256 (5): 274–80. doi:10.1007/s00406-006-0661-7. PMID16788768.
Jump up^ de Boer, S. P.; E. J. Driessen; H. L. Verhaar (1982). Biographical Dictionary of Dissidents in the Soviet Union, 1956-1975. The Hague: Martinus Nijhoff Publishers.ISBN90-247-2538-0.[page needed]
The U.S. Food and Drug Administration today approved a new use for Avastin (bevacizumab) to treat patients with persistent, recurrent or late-stage (metastatic) cervical cancer.
Cervical cancer grows in the tissues of the lower part of the uterus known as the cervix. It commonly occurs when human papillomaviruses (HPV), a virus that spreads through sexual contact, cause cells to become cancerous. Although there are two licensed vaccines available to prevent many types of HPV that can cause cervical cancer, the National Cancer Institute estimates that 12,360 American women will be diagnosed with cervical cancer and 4,020 will die from the disease in 2014.
Avastin works by interfering with the blood vessels that fuel the development of cancerous cells. The new indication for cervical cancer is approved for use in combination with chemotherapy drugs paclitaxel and cisplatin or in combination with paclitaxel and topotecan.
“Avastin is the first drug approved for patients with late-stage cervical cancer since the 2006 approval of topotecan with cisplatin,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “It is also the first biologic agent approved for patients with late-stage cervical cancer and was approved in less than four months under the FDA’s priority review program, demonstrating the agency’s commitment to making promising therapies available to patients faster.”
The FDA reviewed Avastin for treatment of patients with cervical cancer under its priority review program because the drug demonstrated the potential to be a significant improvement in safety or effectiveness over available therapy in the treatment of a serious condition. Priority review provides an expedited review of a drug’s application.
The safety and effectiveness of Avastin for treatment of patients with cervical cancer was evaluated in a clinical study involving 452 participants with persistent, recurrent, or late-stage disease. Participants were randomly assigned to receive paclitaxel and cisplatin with or without Avastin or paclitaxel and topotecan with or without Avastin. Results showed an increase in overall survival to 16.8 months in participants who received chemotherapy in combination with Avastin as compared to 12.9 months for those receiving chemotherapy alone.
The most common side effects associated with use of Avastin in patients with cervical cancer include fatigue, decreased appetite, high blood pressure (hypertension), increased glucose in the blood (hyperglycemia), decreased magnesium in the blood (hypomagnesemia), urinary tract infection, headache and decreased weight. Perforations of the gastrointestinal tract and abnormal openings between the gastrointestinal tract and vagina (enterovaginal fistula) also were observed in Avastin-treated patients.
Avastin is marketed by South San Francisco, California-based Genentech, a member of the Roche Group.
Country
Patent Number
Approved
Expires (estimated)
Canada
2286330
2008-06-10
2018-04-03
Canada
2145985
2003-09-16
2012-10-28
Property
Value
Source
melting point
61 °C (FAB fragment), 71 °C (whole mAb)
Vermeer, A.W.P. & Norde, W., Biophys. J. 78:394-404 (2000)
Protein chemical formula
C6538H10034N1716O2033S44
Protein average weight
149 kDa
A recombinant humanized monoclonal IgG1 antibody that binds to and inhibits the biologic activity of human vascular endothelial growth factor (VEGF). Bevacizumab contains human framework regions and the complementarity-determining regions of a murine antibody that binds to VEGF. Bevacizumab is produced in a Chinese Hamster Ovary mammalian cell expression system in a nutrient medium containing the antibiotic gentamicin and has a molecular weight of approximately 149 kilodaltons.
This is the third portion of the discussion in a series of articles that began with signaling and signaling pathways. There are fine features on the functioning of enzymes and proteins, on sequential changes in a chain reaction, and on conformational changes that we shall return to. These are critical to developing a more complete understanding of life processes. I have indicated that many of the protein-protein interactions or protein-membrane interactions and associated regulatory features have been referred to previously, but the focus of the discussion or points made were different. Even though I considered placing this after the discussion of proteins and how they play out their essential role, I needed to lay out the scope of metabolic reactions and pathways, and their complementary changes. These may not appear to be adaptive, if the…
Percent Composition: C 61.13%, H 4.49%, N 8.91%, O 15.27%, S 10.20%
Properties: Crystals, mp 155-157°. Soly at 25°(mg/ml): water 10 (pH 7.0). Sol in methanol, ethanol; freely sol in organic solvents and alkaline (pH = 12) aqueous solns.
Valdecoxib was manufactured and marketed under the brand name Bextra by G. D. Searle & Company. It was approved by the United States Food and Drug Administration on November 20, 2001,[1] and was available by prescription in tablet form until 2005, when it was removed from the market due to concerns about possible increased risk of heart attack and stroke. The prodrugparecoxib is available in many countries.
Valdecoxib was also used off-label for controlling acute pain and various types of surgical pain.[2]
Side-effects and withdrawal from market
On April 7, 2005, Pfizer withdrew Bextra from the U.S. market on recommendation by the FDA, citing an increased risk of heart attackand stroke and also the risk of a serious, sometimes fatal, skin reaction. This was a result of recent attention to prescription NSAIDs, such as Merck’sVioxx. Other reported side-effects were angina and Stevens–Johnson syndrome.
Pfizer first acknowledged cardiovascular risks associated with Bextra in October 2004. The American Heart Association soon after was presented with a report indicating patients using Bextra while recovering from heart surgery were 2.19 times more likely to suffer a stroke or heart attack than those taking placebos.
In a large study published in JAMA 2006, valdecoxib appeared less adverse for renal (kidney) disease and heart arrhythmia compared to Vioxx, however elevated renal risks were slightly suggested.[3]
2009 settlement for off-label uses promotions
An editor has expressed a concern that this section lends undue weight to certain ideas relative to the article as a whole. Please help to discuss and resolve the dispute before removing this message. (December 2012)
On September 2, 2009, the United States Department of Justice fined Pfizer $2.3 billion after one of its subsidiaries, Pharmacia & UpJohn Company, pled guilty to marketing four drugs including Bextra “with the intent to defraud or mislead.”[4] Pharmacia & UpJohn admitted to criminal conduct in the promotion of Bextra, and agreed to pay the largest criminal fine ever imposed in the United States for any matter, $1.195 billion.[5] A former Pfizer district sales manager was indicted and sentenced to home confinement for destroying documents regarding the illegal promotion of Bextra.[6][7] In addition, a Regional Manager pled guilty to distribution of a mis-branded product, and was fined $75,000 and twenty-four months on probation.[8]
The remaining $1 billion of the fine was paid to resolve allegations under the civil False Claims Act case and is the largest civil fraud settlement against a pharmaceutical company. Six whistle-blowers were awarded more than $102 million for their role in the investigation.[9] Former Pfizer sales representative John Kopchinski acted as a qui tam relator and filed a complaint in 2004 outlining the illegal conduct in the marketing of Bextra.[10] Kopchinski was awarded $51.5 million for his role in the case because the improper marketing of Bextra was the largest piece of the settlement at $1.8 billion.[11]
Several HPLC-UV methods have been reported for valdecoxib estimation in biological samples like human urine,[14] plasma,.[15][16] Valdecoxib has analytical methods for bioequivalence studies,[17][18] metabolite determination,[19][20][21] and estimation of formulation,[22] HPTLC method for simultaneous estimation in tablet dosage form.[23]
Deoxybenzoin (I) is converted to the corresponding oxime (II) by treatment with NH2OH稨Cl under basic conditions either with sodium acetate in aqueous ethanol or in toluene in presence of potassium hydroxide in absolute ethanol. Deprotonation of the oxime under nitrogen with 2eq of butyllithium in THF followed by cyclization in ethyl acetate or acetic anhydride affords isoxazoline (III). Finally, treatment of (III) with cold chlorosulfonic acid followed by reaction of the intermediate sulfonyl chloride with aqueous ammonia affords the desired product.
J Med Chem2000,43,(5):775
Trade Names
Country
Trade name
Manufacturer
Germany
Bextra
Pharmacia
USA
– “-
– “-
Ukraine
No
No
Formulations
Tablets of 10 mg, 20 mg
Valdecoxib is chemically designated as 4-(5-methyl-3-phenyl-4-isoxazolyl) benzenesulfonamide and is a diaryl substituted isoxazole.
The empirical formula for valdecoxib is C16H14N2O3S, and the molecular weight is 314.36. Valdecoxib is a white crystalline powder that is relatively insoluble in water (10 µg/mL) at 25° C and pH 7.0, soluble in methanol and ethanol, and freely soluble in organic solvents and alkaline (pH=12) aqueous solutions.
BEXTRA (valdecoxib) Tablets for oral administration contain either 10 mg or 20 mg of valdecoxib. Inactive ingredients include lactose monohydrate, microcrystalline cellulose, pregelatinized starch, croscarmellose sodium,magnesium stearate, hydroxypropyl methylcellulose, polyethylene glycol, polysorbate 80, and titanium dioxide
………………………
NMR
Links
Talley, JJ et al .: J. Med. Chem. (JMCMAR) 43, 775-777 (2000).
US 5,859,257 (GD Searle; 12.1.1999; USA-prior. 13.2.1995).
Literature References:
Selective cyclooxygenase-2 (COX-2) inhibitor. Active metabolite of parecoxib, q.v. Prepn: J. J. Talley et al.,WO9625405 (1996 to Searle); eidem, US5633272 (1997); and activity: eidem, J. Med. Chem.43, 775 (2000).
Chromatographic determn of purity: D. A. Roston et al., J. Pharm. Biomed. Anal.26, 339 (2001).
Gastrointestinal tolerability study: G. M. Eisen et al.,Aliment. Pharmacol. Ther.21, 591 (2005).
Clinical trial in hip arthroplasty: F. Camu et al., Am. J. Ther.9, 43 (2002).
Clinical comparison with oxycodone/acetominophen in dental pain: S. E. Daniels et al., J. Am. Dent. Assoc.133, 611 (2002).
Clinical trial in migraine: D. Kudrow et al.,Headache45, 1151 (2005).
Review of clinical experience: M. Goldman, S. Schutzer, Formulary37, 68-77 (2002); of clinical efficacy and safety: G. P. Joshi, Expert Rev. Neurother.5, 11-24 (2005).
Jump up^Talley, J. J.; Brown, D. L.; Carter, J. S.; Graneto, M. J.; Koboldt, C. M.; Masferrer, J. L.; Perkins, W. E.; Rogers, R. S.; Shaffer, A. F.; Zhang, Y. Y.; Zweifel, B. S.; Seibert, K. (2000). “4-[5-Methyl-3-phenylisoxazol-4-yl]- benzenesulfonamide, Valdecoxib: A Potent and Selective Inhibitor of COX-2”. Journal of Medicinal Chemistry43 (5): 775–777. doi:10.1021/jm990577v.PMID10715145.edit
Jump up^Prafulla Kumar Sahu and M. Mathrusri Annapurna, Analytical method development by liquid chromatography, LAP Lambert Academic Publisher, Germany, 2011 ISBN 3-8443-2869-6.
Jump up^Zhang J Y, Fast D M and Breau A P, J Chromatogr B Analyt Technol Biomed Life Sci., 2003, 785(1), 123-134
Jump up^Ramakrishna N V S, Vishwottam K N; Wishu S and Koteshwara M, J Chromatogr B Analyt Technol Biomed Life Sci., 2004, 802(2), 271.
Jump up^Sane R T, Menon S, Deshpande A Y and Jain A, Chromatogr., 2005, 61(3-4), 137-141.
Jump up^Prafulla Kumar Sahu*, K. Ravi Sankar and M. Mathrusri Annapurna, Determination of Valdecoxib in human plasma using Reverse Phase HPLC”, E-Journal of Chemistry, 2011, 8(2), 875-881.
Jump up^Mandal U, Jayakumar M, Ganesan M, Nandi S, Pal T K, Chakraborty M K, Roy Chowdhary A. and Chattoraj T K, Indian Drugs, 2004, 41, 59.
Zhang J.Y, Fast D.M and Breau, A.P, J Pharm Biomed Anal., 2003, 33, 61.
Werner U, Werner D, Hinz B, Lanbrecht C and Brune K, J Biomed Chromatogr., 2004, 19, 113.
Zhang J V, Fast D M and Breau A P, J Chromatogr B Anal Technol Biomed Life Sci., 2003, 785, 123.
Sutariya V B, Rajashree M, Sankalia M G. and Priti P, Indian J Pharm Sci., 2004, 93, 112.
JGandhimathi M, Ravi T K, Shukla Nilima and Sowmiya G, Indian J Pharm Sci., 2007, 69(1), 145-147.
Abacavir (ABC) is a powerful nucleoside analog reverse transcriptase inhibitor (NRTI) used to treat HIV and AIDS. [Wikipedia] Chemically, it is a synthetic carbocyclic nucleoside and is the enantiomer with 1S, 4R absolute configuration on the cyclopentene ring. In vivo, abacavir sulfate dissociates to its free base, abacavir.
Abacavir (ABC) i/ʌ.bæk.ʌ.vɪər/ is a nucleoside analog reverse transcriptase inhibitor (NRTI) used to treat HIV and AIDS. It is available under the trade name Ziagen (ViiV Healthcare) and in the combination formulations Trizivir (abacavir, zidovudine andlamivudine) and Kivexa/Epzicom (abacavir and lamivudine). It has been well tolerated: the main side effect is hypersensitivity, which can be severe, and in rare cases, fatal. Genetic testing can indicate whether an individual will be hypersensitive; over 90% of patients can safely take abacavir. However, in a separate study, the risk of heart attack increased by nearly 90%.[1]
Viral strains that are resistant to zidovudine (AZT) orlamivudine (3TC) are generally sensitive to abacavir (ABC), whereas some strains that are resistant to AZT and 3TC are not as sensitive to abacavir.
Abacavir is a nucleoside reverse transcriptase inhibitor (NRTI) with activity against Human Immunodeficiency Virus Type 1 (HIV-1). Abacavir is phosphorylated to active metabolites that compete for incorporation into viral DNA. They inhibit the HIV reverse transcriptase enzyme competitively and act as a chain terminator of DNA synthesis. The concentration of drug necessary to effect viral replication by 50 percent (EC50) ranged from 3.7 to 5.8 μM (1 μM = 0.28 mcg/mL) and 0.07 to 1.0 μM against HIV-1IIIB and HIV-1BaL, respectively, and was 0.26 ± 0.18 μM against 8 clinical isolates. Abacavir had synergistic activity in cell culture in combination with the nucleoside reverse transcriptase inhibitor (NRTI) zidovudine, the non-nucleoside reverse transcriptase inhibitor (NNRTI) nevirapine, and the protease inhibitor (PI) amprenavir; and additive activity in combination with the NRTIs didanosine, emtricitabine, lamivudine, stavudine, tenofovir, and zalcitabine.
Abacavir is a carbocyclic synthetic nucleoside analogue and an antiviral agent. Intracellularly, abacavir is converted by cellular enzymes to the active metabolite carbovir triphosphate, an analogue of deoxyguanosine-5′-triphosphate (dGTP). Carbovir triphosphate inhibits the activity of HIV-1 reverse transcriptase (RT) both by competing with the natural substrate dGTP and by its incorporation into viral DNA. Viral DNA growth is terminated because the incorporated nucleotide lacks a 3′-OH group, which is needed to form the 5′ to 3′ phosphodiester linkage essential for DNA chain elongation.
Application
an antiviral agent, is used in the treatment of AIDS
Abacavir, (-) cis-[4-[2-amino-6-cyclopropylamino)-9H-purin-9-yl]-2-cyclopenten-yl]-1 – methanol, a carbocyclic nucleoside which possesses a 2,3-dehydrocyclopentene ring, is referred to in United States Patent 5,034,394 as a reverse transcriptase inhibitor. Recently, a general synthetic strategy for the preparation of this type of compound and intermediates was reported [Crimmins, et. al., J. Org. Chem., 61 , 4192-4193 (1996) and 65, 8499-8509-4193 (2000)].
Abacavir is the International Nonproprietary Name (INN) of {(1S,4R)-4-[2-amino-6-(cyclopropylamino)-9H-purin-9-yl]-cyclopent-2-enyl}methanol and CAS No. 136470-78-5. Abacavir and therapeutically acceptable salts thereof, in particular the hemisulfate salt, are well-known as potent selective inhibitors of HIV-1 and HIV-2, and can be used in the treatment of human immunodeficiency virus (HIV) infection.
The structure of abacavir corresponds to formula (I):
EP 434450-A discloses certain 9-substituted-2-aminopurines including abacavir and its salts, methods for their preparation, and pharmaceutical compositions using these compounds.
Different preparation processes of abacavir are known in the art. In some of them abacavir is obtained starting from an appropriate pyrimidine compound, coupling it with a sugar analogue residue, followed by a cyclisation to form the imidazole ring and a final introduction of the cyclopropylamino group at the 6 position of the purine ring.
According to the teachings of EP 434450-A , the abacavir base is finally isolated by trituration using acetonitrile (ACN) or by chromatography, and subsequently it can be transformed to a salt of abacavir by reaction with the corresponding acid. Such isolation methods (trituration and chromatography) usually are limited to laboratory scale because they are not appropriate for industrial use. Furthermore, the isolation of the abacavir base by trituration using acetonitrile gives a gummy solid (Example 7) and the isolation by chromatography (eluted from methanol/ethyl acetate) yields a solid foam (Example 19 or 28).
Other documents also describe the isolation of abacavir by trituration or chromatography, but always a gummy solid or solid foam is obtained (cf. WO9921861 and EP741710 ), which would be difficult to operate on industrial scale.
WO9852949 describes the preparation of abacavir which is isolated from acetone. According to this document the manufacture of the abacavir free base produces an amorphous solid which traps solvents and is, therefore, unsuitable for large scale purification, or for formulation, without additional purification procedures (cf. page 1 of WO 9852949 ). In this document, it is proposed the use of a salt of abacavir, in particular the hemisulfate salt which shows improved physical properties regarding the abacavir base known in the art. Said properties allow the manufacture of the salt on industrial scale, and in particular its use for the preparation of pharmaceutical formulations.
However, the preparation of a salt of abacavir involves an extra processing step of preparing the salt, increasing the cost and the time to manufacture the compound. Generally, the abacavir free base is the precursor compound for the preparation of the salt. Thus, depending on the preparation process used for the preparation of the salt, the isolation step of the abacavir free base must also be done.
EXAMPLE 21(-)-cis-4-[2-Amino-6-(cyclopropylmethylamino)-9H-purin-9-yl]-2-cyclopentene-1-methanol
The title compound of Example 7, (2.00 g, 6.50 mmol) was dissolved in 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (Aldrich, 20 mL). Phosphoryl chloride (2.28 mL, 24.0 mmol) was added to the stirred, cooled (-10° C.) solution. After 3 minutes, cold water (80 mL) was added. The solution was extracted with chloroform (3×80 mL). The aqueous layer was diluted with ethanol (400 mL) and the pH adjusted to 6 with saturated aqueous NaOH. The precipitated inorganic salts were filtered off. The filtrate was further diluted with ethanol to a volume of 1 liter and the pH adjusted to 8 with additional NaOH. The resulting precipitate was filtered and dried to give the 5′-monophosphate of (±)-cis-4-[2-amino-6-(cyclopropylmethylamino)-9H-purin-9-yl]-2-cyclopentene-1-methanol as white powder (4.0 mmoles, 62% quantitated by UV absorbance); HPLC analysis as in Example 17 shows one peak. This racemic 5′ -monophosphate was dissolved in water (200 mL) and snake venom 5′-nucleotidase (EC 3.1.3.5) from Crotalus atrox (5,000 IU, Sigma) was added. After incubation at 37° C. for 10 days, HPLC analysis as in Example 17 showed that 50% of the starting nucleotide had been dephosphorylated to the nucleoside. These were separated on a 5×14 cm column of DEAE Sephadex A25 (Pharmacia) which had been preequilibrated with 50 mM ammonium bicarbonate. Title compound was eluted with 2 liters of 50 mM ammonium bicarbonate. Evaporation of water gave white powder which was dissolved in methanol, adsorbed on silica gel, and applied to a silica gel column. Title compound was eluted with methanol:chloroform/1:9 as a colorless glass. An acetonitrile solution was evaporated to give white solid foam, dried at 0.3 mm Hg over P2 O5 ; 649 mg (72% from racemate); 1 H-NMR in DMSO-d6 and mass spectrum identical with those of the racemate (title compound of Example 7); [α]20D -48.0°, [α]20436 -97.1°, [α]20365 -149° (c=0.14, methanol).
Anal. Calcd. for C15 H20 N6 O.0.10CH3 CN: C, 59.96; H, 6.72; N, 28.06. Found: C, 59.93; H, 6.76; N, 28.03.
Continued elution of the Sephadex column with 2 liters of 100 mM ammonium bicarbonate and then with 2 liters of 200 mM ammonium bicarbonate gave 5′-monophosphate (see Example 22) which was stable to 5′-nucleotidase.
…………………………………………
Синтез a)
Синтез b)
Preparation c)
Synthesis d)
An enantiopure β-lactam with a suitably disposed electron withdrawing group on nitrogen, participated in a π-allylpalladium mediated reaction with 2,6-dichloropurine tetrabutylammonium salt to afford an advanced cis-1,4-substituted cyclopentenoid with both high regio- and stereoselectivity. This advanced intermediate was successfully manipulated to the total synthesis of (−)-Abacavir.
Example 1: Preparation of crystalline Form I of abacavir base using methanol as solvent
[0026]
Abacavir (1.00 g, containing about 17% of dichloromethane) was dissolved in refluxing methanol (2.2 mL). The solution was slowly cooled to – 5 °C and, the resulting suspension, was kept at that temperature overnight under gentle stirring. The mixture was filtered off and dried under vacuum (7-10 mbar) at 40 °C for 4 hours to give a white solid (0.55 g, 66% yield, < 5000 ppm of methanol). The PXRD analysis gave the diffractogram shown in FIG. 1.
Abacavir, is the International Nonproprietary Name (INN) of {(1 S,4R)-4-[2- amino-6-(cyclopropylamino)-9H-purin-9-yl]-cyclopent-2-enyl}methanol, and CAS No. 136470-78-5. Abacavir sulfate is a potent selective inhibitor of HIV-1 and HIV-2, and can be used in the treatment of human immunodeficiency virus (HIV) infection.
The structure of abacavir hemisulfate salt corresponds to formula (I):
(I)
EP 434450-A discloses certain 9-substituted-2-aminopuhnes including abacavir and its salts, methods for their preparation, and pharmaceutical compositions using these compounds.
Different preparation processes of abacavir are known in the art. In some of them abacavir is obtained starting from an appropriate pyrimidine compound, coupling it with a sugar analogue residue, followed by a cyclisation to form the imidazole ring and a final introduction of the cyclopropylamino group at the 6 position of the purine ring. Pyrimidine compounds which have been identified as being useful as intermediates of said preparation processes include N-2-acylated abacavir intermediates such as N-{6- (cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent-2-enyl]-9H-purin- 2-yl}acetamide or N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-
(hydroxymethyl)cyclopent-2-enyl]-9H-purin-2-yl}isobutyramide. The removal of the amino protective group of these compounds using acidic conditions is known in the art. According to Example 28 of EP 434450-A, the amino protective group of the N-{6-(cyclopropylamino)-9-[(1 R,4S)-4- (hydroxymethyl)cyclopent-2-enyl]-9H-purin-2-yl}isobutyramide is removed by stirring with 1 N hydrochloric acid for 2 days at room temperature. The abacavir base, after adjusting the pH to 7.0 and evaporation of the solvent, is finally isolated by trituration and chromatography. Then, it is transformed by reaction with an acid to the corresponding salt of abacavir. The main disadvantages of this method are: (i) the use of a strongly corrosive mineral acid to remove the amino protective group; (ii) the need of a high dilution rate; (iii) a long reaction time to complete the reaction; (iv) the need of isolating the free abacavir; and (v) a complicated chromatographic purification process.
Thus, despite the teaching of this prior art document, the research of new deprotection processes of a N-acylated {(1 S,4R)-4-[2-amino-6- (cyclopropylamino)-9H-purin-9-yl]-cyclopent-2-enyl}methanol is still an active field, since the industrial exploitation of the known process is difficult, as it has pointed out above. Thus, the provision of a new process for the removal of the amino protective group of a N-acylated {(1 S,4R)-4-[2-amino-6-
(cyclopropylamino)-9H-purin-9-yl]-cyclopent-2-enyl}methanol is desirable.
Example 1 : Preparation of abacavir hemisulfate
N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent-2-enyl]-9H- purin-2-yl}isobutyramide (6.56 g, 18.40 mmol) was slurried in a mixture of isopropanol (32.8 ml) and 10% solution of NaOH (36.1 ml, 92.0 mmol). The mixture was refluxed for 1 h. The resulting solution was cooled to 20-25 0C and tert-butyl methyl ether (32.8 ml) was added. The layers were separated and H2SO4 96% (0.61 ml, 11.03 mmol) was added dropwise to the organic layer. This mixture was cooled to 0-50C and the resulting slurry filtered off.
The solid was dried under vacuum at 40 0C. Abacavir hemisulfate (5.98 g, 97%) was obtained as a white powder.
Example 6: Preparation of abacavir
N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent-2-enyl]-9H- purin-2-yl}isobutyramide (1.0 g, 2.80 mmol) was slurried in a mixture of isopropanol (2 ml) and 10% solution of NaOH (1.1 ml, 2.80 mmol). The mixture was refluxed for 1 h. The resulting solution was cooled to 20-25 0C and tert-butyl methyl ether (2 ml) was added. The aqueous layer was discarded, the organic phase was cooled to 0-5 0C and the resulting slurry filtered off. The solid was dried under vacuum at 400C. Abacavir (0.62 g, 77%) was obtained as a white powder.
Example 7: Preparation of abacavir
N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent-2-enyl]-9H- purin-2-yl}isobutyramide (1.25 g, 3.51 mmol) was slurried in a mixture of isopropanol (2.5 ml) and 10% solution of NaOH (1.37 ml, 3.51 mmol). The mixture was refluxed for 1 h and concentrated to dryness. The residue was crystallized in acetone. Abacavir (0.47 g, 47%) was obtained as a white powder.
Example 8: Preparation of abacavir
N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent-2-enyl]-9H- purin-2-yl}isobutyramide (1.25 g, 3.51 mmol) was slurried in a mixture of isopropanol (2.5 ml) and 10% solution of NaOH (1.37 ml, 3.51 mmol). The mixture was refluxed for 1 h and concentrated to dryness. The residue was crystallized in acetonitrile. Abacavir (0.43 g, 43%) was obtained as a white powder.
Example 9: Preparation of abacavir
A mixture of N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent- 2-enyl]-9H-purin-2-yl}isobutyramide (10 g, 28 mmol), isopropanol (100 ml) and 10% solution of NaOH (16.8 ml, 42 mmol) was refluxed for 1 h. The resulting solution was cooled to 20-25 0C and washed several times with 25% solution of NaOH (10 ml). The wet organic layer was neutralized to pH 7.0-7.5 with 17% hydrochloric acid and it was concentrated to dryness under vacuum. The residue was crystallized in ethyl acetate (150 ml) to afford abacavir (7.2 g, 90%).
Example 10: Preparation of abacavir
A mixture of N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent- 2-enyl]-9H-purin-2-yl}isobutyramide (10 g, 28 mmol), isopropanol (100 ml) and 10% solution of NaOH (16.8 ml, 42 mmol) was refluxed for 1 h. The resulting solution was cooled to 20-25 0C and washed several times with 25% solution of NaOH (10 ml). The wet organic layer was neutralized to pH 7.0-7.5 with 17% hydrochloric acid and it was concentrated to dryness under vacuum. The residue was crystallized in acetone (300 ml) to afford abacavir (7.0 g, 88%).
or (1 S,4R)-4-[2-Amino-6-(cyclopropylamino)-9H-purin-9-yl]-2-cyclopentene-1 – methanol and its salts are nucleoside reverse transcriptase inhibitors. Abacavir sulfate is a nucleoside reverse transcriptase inhibitor and used in the treatment of human immunodeficiency virus infection. Abacavir sulfate and related compounds and their therapeutic uses are disclosed in US 5,034,394.
Crystalline forms of abacavir sulfate have not been reported in the literature. Moreover, the processes described in the literature do not produce abacavir sulfate in a stable, well-defined and reproducible crystalline form. It has now been discovered that abacavir sulfate can be prepared in three stable, well-defined and consistently reproducible crystalline forms.
Example 1
Abacavir free base (3.0 gm, obtained by the process described in example 21 of US 5,034,394) is dissolved in ethyl acetate (15 ml) and cone, sulfuric acid (0.3 ml) is added to the solution. Then the contents are stirred for 3 hours at 20°C and filtered to give 3.0 gm of form I abacavir sulfate. Example 2 Abacavir free base (3.0 gm) is dissolved in acetone (20 ml) and cone, sulfuric acid (0.3 ml) is added to the solution. Then the contents are stirred for 6 hours at 25°C and filtered to give 2.8 gm of form I abacavir sulfate.
Example 3 Abacavir free base (3.0 gm) is dissolved in acetonitrile (15 ml) and sulfuric acid (0.3 ml) is added to the solution. Then the contents are stirred for 2 hours at 25°C and the separated solid is filtered to give 3.0 gm of form II abacavir sulfate.
Example 4 Abacavir free base (3.0 gm) is dissolved in methyl tert-butyl ether (25 ml) and sulfuric acid (0.3 ml) is added to the solution. Then the contents are stirred for 1 hours at 25°C and the separated solid is filtered to give 3.0 gm of form II abacavir sulfate.
Example 5 Abacavir free base (3.0 gm) is dissolved in methanol (15 ml) and sulfuric acid (0.3 ml) is added to the solution. The contents then are cooled to 0°C and diisopropyl ether (15 ml) is added. The reaction mass is stirred for 2 hours at about 25°C and the separated solid is filtered to give 3.0 gm of form III abacavir sulfate
The present invention relates to a new process for the preparation of the chiral nucleoside analogue (1S, 4R)-4-[2-amino-6-(cyclopropylamino)-9H purin-9-yl]-2- cyclopentene-1 -methanol (compound of Formula (I)).
The compound of formula (I) is described as having potent activity against human immunodeficiency virus (HIV) and hepatitis B virus (HBV) in EPO34450.
Results presented at the 34th Interscience Conference on Antimicrobial Agents and Chemotherapy (October 4-7, 1994) demonstrate that the compound of formula I has significant activity against HIV comparable to, and if not better than, some current anti HIV drugs, such as zidovudine and didanosine.
Currently the compound of Formula (I) is undergoing clinical investigation to determine its safety and efficacy in humans. Therefore, there exists at the present time a need to supply large quantities of this compound for use in clinical trials.
Current routes of synthesising the compound of formula (I) involve multiple steps and are relatively expensive. It will be noted that the compound has two centres of asymmetry and it is essential that any route produces the compound of formula (I) substantially free of the corresponding enantiomer, preferably the compound of formula (I) is greater than 95% w/w free of the corresponding enantiomer.
Processes proposed for the preparation of the compound of formula (I) generally start from a pyrimidine compound, coupling with a 4-amino-2-cyclopentene-1- methanol analogue, cyciisation to form the imidazole ring and then introduction of the cyclopropylamine group into the 6 position of the purine, such routes include those suggested in EPO434450 and WO9521161. Essentially both routes disclosed in the two prior patent applications involve the following steps:-
(i) coupling (1S, 4R)-4-amino-2-cyclopentene-1 -methanol to N-(4,6-dichloro-5- formamido-2-pyrimidinyl) acetamide or a similar analogue thereof, for example N- (2-amino-4,6-dichloro-5-pyrimidinyl) formamide;
(ii) ring closure of the resultant compound to form the intermediate (1 S, 4R)-4- (2-amino-6-chloro-9H-purin-9-yl)-2-cyclopentene-1 -methanol;
(iii) substituting the halo group by a cyclopropylamino group on the 6 position of the purine ring.
The above routes are multi-step processes. By reducing the number of processing steps significant cost savings can be achieved due to the length of time to manufacture the compound being shortened and the waste streams minimised.
An alternative process suggested in the prior art involves the direct coupling of carbocyclic ribose analogues to the N atom on the 9 position of 2-amino-6-chloro purine. For example WO91/15490 discloses a single step process for the formation of the (1S, 4R)- 4-(2-amino-6-chloro-9H-purin-9-yl)-2-cyclopentene-1- methanol intermediate by reacting (1S, 4R)-4-hydroxy-2-cyclopentene-1 -methanol, in which the allylic hydroxyl group has been activated as an ester or carbonate and the other hydroxyl group has a blocking group attached (for example 1 ,4- bis- methylcarbonate) with 2-amino-6-chloropurine.
However we have found that when synthesising (1S, 4R)-4-(2-amino-6-chloro-9H- purin-9-yl)-2-cyclopentene-1- methanol by this route a significant amount of an N- 7 isomer is formed (i.e. coupling has occurred to the nitrogen at the 7- position of the purine ring) compared to the N-9 isomer desired. Further steps are therefore required to convert the N-7 product to the N-9 product, or alternatively removing the N-7 product, adding significantly to the cost. We have found that by using a transition metal catalysed process for the direct coupling of a compound of formula (II) or (III),
Example 1 (1 S. 4R)-4-[2-Amino-6-(cvclopropylamino)-9H purin-9-vπ-2-cvclopentene-1 – methanol
Triphenylphosphine (14mg) was added, under nitrogen, to a mixture of (1S.4R)- 4-hydroxy-2-cyclopentene -1 -methanol bis(methylcarbonate) (91 mg), 2-amino-6- (cyclopropylamino) purine (90mg), tris(dibenzylideneacetone)dipalladium (12mg) and dry DMF (2ml) and the resulting solution stirred at room temperature for 40 min.
The DMF was removed at 60° in vacuo and the residue partitioned between ethyl acetate (25ml.) and 20% sodium chloride solution (10ml.). The ethyl acetate solution was washed with 20% sodium chloride (2x12ml.) and with saturated sodium chloride solution, then dried (MgSO4) and the solvent removed in vacuo.
The residue was dissolved in methanol (10ml.), potassium carbonate (17mg) added and the mixture stirred under nitrogen for 15h.
The solvent was removed in vacuo and the residue chromatographed on silica gel
(Merck 9385), eluting with dichloromethane-methanol [(95:5) increasing to (90:10)] to give the title compound (53mg) as a cream foam.
Trizivir tablets 300 mg – abacavir in fixed combination with 150 mg of lamivudine and 300 mg zidovudine
ZIAGEN is the brand name for abacavir sulfate, a synthetic carbocyclic nucleoside analogue with inhibitory activity against HIV-1. The chemical name of abacavir sulfate is (1S,cis)-4-[2-amino-6-(cyclopropylamino)-9H-purin-9-yl]-2-cyclopentene-1-methanol sulfate (salt) (2:1). Abacavir sulfate is the enantiomer with 1S, 4R absolute configuration on the cyclopentene ring. It has a molecular formula of (C14H18N6O)2•H2SO4 and a molecular weight of 670.76 daltons. It has the following structural formula:
Abacavir sulfate is a white to off-white solid with a solubility of approximately 77 mg/mL in distilled water at 25°C. It has an octanol/water (pH 7.1 to 7.3) partition coefficient (log P) of approximately 1.20 at 25°C.
ZIAGEN Tablets are for oral administration. Each tablet contains abacavir sulfate equivalent to 300 mg of abacavir as active ingredient and the following inactive ingredients: colloidal silicon dioxide, magnesium stearate, microcrystalline cellulose, and sodium starch glycolate. The tablets are coated with a film that is made of hypromellose, polysorbate 80, synthetic yellow iron oxide, titanium dioxide, and triacetin.
ZIAGEN Oral Solution is for oral administration. Each milliliter (1 mL) of ZIAGEN Oral Solution contains abacavir sulfate equivalent to 20 mg of abacavir (i.e., 20 mg/mL) as active ingredient and the following inactive ingredients: artificial strawberry and banana flavors, citric acid (anhydrous), methylparaben and propylparaben (added as preservatives), propylene glycol, saccharin sodium, sodium citrate (dihydrate), sorbitol solution, and water.
In vivo, abacavir sulfate dissociates to its free base, abacavir. All dosages for ZIAGEN are expressed in terms of abacavir.
History
Abacavir was approved by the Food and Drug Administration (FDA) on December 18, 1998 and is thus the fifteenth approved antiretroviral drug in the United States. Its patent expired in the United States on 2009-12-26.
Links
US 5 089 500 (Burroughs Wellcome; 18.2.1992; GB-prior. 27.6.1988).
Synthesis a)
EP 434 450 (Wellcome Found .; 26.6.1991; appl. 21.12.1990; prior-USA. 22.12.1989).
Crimmins, MT et al .: J. Org. Chem. (JOCEAH) 61 4192 (1996).
EP 1 857 458 (Solmag; appl. 5.5.2006).
EP 424 064 (Enzymatix; appl. 24.4.1991; GB -prior. 16.10.1989).
Jump up^Mallal, S., Phillips, E., Carosi, G. et al. (2008). “HLA-B*5701 screening for hypersensitivity to abacavir”. New England Journal of Medicine358: 568–579.doi:10.1056/nejmoa0706135.
Jump up^Rauch, A., Nolan, D., Martin, A. et al. (2006). “Prospective genetic screening decreases the incidence of abacavir hypersensitivity reactions in the Western Australian HIV cohort study”. Clinical Infectious Diseases43: 99–102. doi:10.1086/504874.
Jump up^Heatherington et al. (2002). “Genetic variations in HLA-B region and hypersensitivity reactions to abacavir”. Lancet359: 1121–1122.
Jump up^Mallal et al. (2002). “Association between presence of HLA*B5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir”. Lancet359: 727–732. doi:10.1016/s0140-6736(02)07873-x.
Jump up^Rotimi, C.N.; Jorde, L.B. (2010). “Ancestry and disease in the age of genomic medicine”. New England Journal of Medicine363: 1551–1558.
Jump up^Phillips, E., Mallal, S. (2009). “Successful translation of pharmacogenetics into the clinic”. Molecular Diagnosis & Therapy13: 1–9. doi:10.1007/bf03256308.
Jump up^Phillips, E., Mallal S. (2007). “Drug hypersensitivity in HIV”. Current Opinion in Allergy and Clinical Immunology7: 324–330. doi:10.1097/aci.0b013e32825ea68a.
Jump up^Swen JJ, Nijenhuis M, de Boer A et al. (May 2011). “Pharmacogenetics: from bench to byte–an update of guidelines”. Clin Pharmacol Ther.89 (5): 662–73.doi:10.1038/clpt.2011.34. PMID21412232.
Jump up^Shear, N.H., Milpied, B., Bruynzeel, D.P. et al. (2008). “A review of drug patch testing and implications for HIV clinicians”. AIDS22: 999–1007.doi:10.1097/qad.0b013e3282f7cb60.
Jump up^Ding X, Andraca-Carrera E, Cooper C et al. (December 2012). “No association of abacavir use with myocardial infarction: findings of an FDA meta-analysis”. J Acquir Immune Defic Syndr.61 (4): 441–7. doi:10.1097/QAI.0b013e31826f993c.PMID22932321.
Illing PT et al. 2012, Nature, doi:10.1038/nature11147
Carbocyclic nucleosides are synthetically the most challenging class of nucleosides, requiring multi-step and often elaborate synthetic pathways to introduce the necessary stereochemistry. There are two main strategies for the preparation of carbocyclic nucleosides. In the linear approach a cyclopentylamine is used as starting material and the heterocycle is built in a stepwise manner (see Scheme 1).
Scheme 1: Linear approach for the synthesis of abacavir.[5]
The more flexible strategy is a convergent approach: a functionalized carbocyclic moiety is condensed with a heterocycle rapidly leading to a variety of carbocyclic nucleosides. Initially, we started our syntheses from cyclopentadiene 1 that is deprotonated and alkylated with benzyloxymethyl chloride to give the diene 2. This material is converted by a hydroboration into cyclopentenol 3 or isomerized into two thermodynamically more stable cyclopentadienes 4a,b. With the protection and another hydroboration step to 5 we gain access to an enantiomerically pure precursor for the synthesis of a variety of carbocyclic 2’-deoxynucleosides e.g.:carba-dT, carba-dA or carba-BVDU.[6] The isomeric dienes 4a,b were hydroborated to the racemic carbocyclic moiety 6.
Scheme 2: Convergent approach for the synthesis of carba-dT.
The asymmetric synthesis route and the racemic route above are short and efficient ways to diverse carbocyclic D- or L-nucleosides (Scheme 2). Different heterocycles can be condensed to these precursors leading to carbocyclic purine- and pyrimidine-nucleosides. Beside α- and β-nucleosides, carbocyclic epi– andiso-nucleosides in the 2’-deoxyxylose form were accessable.[7]
What else is possible? The racemic cyclopentenol 6 can be coupled by a modified Mitsunobu-reaction.Moreover, this strategy offers the possibility of synthesizing new carbocyclic nucleosides by functionalizing the double bond before or after introduction of the nucleobase (scheme 3).[8]
Scheme 3: Functionalized carbocyclic nucleosides based on cyclopentenol 6.
Other interesting carbocyclic precursors like cyclopentenol 7 can be used to synthesize several classes of carbocyclic nucleoside analogues, e.g.: 2’,3’-dideoxy-2’,3’-didehydro nucleosides (d4-nucleosides), 2’,3’-dideoxynucleosides (ddNs), ribonucleosides, bicyclic nucleosides or even 2’-fluoro-nucleosides.
Scheme 4: Functionalized carbocyclic thymidine analogues based on cyclopentenol 7.
[1] V. E. Marquez, T. Ben-Kasus, J. J. Barchi, K. M. Green, M .C. Nicklaus, R. Agbaria, J. Am. Chem. Soc.2004,126, 543.
[2] A. D. Borthwick, K. Biggadike, Tetrahedron1992, 48, 571.
[3] H. Bricaud, P. Herdewijn, E. De Clercq, Biochem. Pharmacol.1983, 3583.
[4] P. L. Boyer, B. C. Vu, Z. Ambrose, J. G. Julias, S. Warnecke, C. Liao, C. Meier, V. E. Marquez, S. H. Hughes, J. Med. Chem.2009, 52, 5356.
[5] S. M. Daluge, M. T. Martin, B. R. Sickles, D. A. Livingston, Nucleosides, Nucleotides Nucleic Acids2000,19, 297.
[6] O. R. Ludek, C. Meier, Synthesis2003, 2101.
[7] O. R. Ludek, T. Kraemer, J. Balzarini, C. Meier, Synthesis2006, 1313.
[8] M. Mahler, B. Reichardt, P. Hartjen, J. van Lunzen, C. Meier, Chem. Eur. J.2012, 18, 11046-11062.
The FDA has approved a new type of sleep drug. This new drug is an orexin receptor antagonist and is the first approved drug of this type. Orexins are chemicals that are involved in regulating the sleep-wake cycle and play a role in keeping people awake. Learn more here:http://go.usa.gov/EcEz
The U.S. Food and Drug Administration today approved Belsomra (suvorexant) tablets for use as needed to treat difficulty in falling and staying asleep (insomnia).
Belsomra is an orexin receptor antagonist and is the first approved drug of this type. Orexins are chemicals that are involved in regulating the sleep-wake cycle and play a role in keeping people awake. Belsomra alters the signaling (action) of orexin in the brain.
Insomnia is a common condition in which a person has trouble falling or staying asleep. It can range from mild to severe, depending on how often it occurs and for how long. Insomnia can cause daytime sleepiness and lack of energy. It also can make a person feel anxious, depressed, or irritable. People with insomnia may have trouble with attentiveness, learning, and memory.
“To assist health care professionals and patients in finding the best dose to treat each individual patient’s sleeplessness, the FDA has approved Belsomra in four different strengths – 5, 10, 15, and 20 milligrams,” said Ellis Unger, M.D., director of the Office of Drug Evaluation I in the FDA’s Center for Drug Evaluation and Research. “Using the lowest effective dose can reduce the risk of side effects, such as next-morning drowsiness.”
Belsomra should be taken no more than once per night, within 30 minutes of going to bed, with at least seven hours remaining before the planned time of waking. The total dose should not exceed 20 mg once daily.
The most commonly reported adverse reaction reported by clinical trial participants taking Belsomra was drowsiness. Medications that treat insomnia can cause next-day drowsiness and impair driving and other activities that require alertness. People can be impaired even when they feel fully awake.
The FDA asked the drug manufacturer, Merck, Sharpe & Dohme Corp., to study next-day driving performance in people who had taken Belsomra. The testing showed impaired driving performance in both male and female participants when the 20 mg strength was taken. Patients using the 20 mg strength should be cautioned against next-day driving or activities requiring full mental alertness. Patients taking lower doses should also be made aware of the potential for next-day driving impairment, because there is individual variation in sensitivity to the drug.
The effectiveness of Belsomra was studied in three clinical trials involving more than 500 participants. In the studies, patients taking the drug fell asleep faster and spent less time awake during the remainder of the night compared to people taking an inactive pill (placebo). Belsomra was not compared to other drugs approved to treat insomnia, so it is not known if there are differences in safety or effectiveness between Belsomra and other insomnia medications.
Like other sleep medicines, there is a risk from Belsomra of sleep-driving and other complex behaviors while not being fully awake, such as preparing and eating food, making phone calls, or having sex. Chances of such activity increase if a person has consumed alcohol or taken other medicines that make them sleepy. Patients or their families should call the prescribing health care professional if this type of activity occurs.
Belsomra will be dispensed with an FDA-approved patient Medication Guide that provides instructions for its use and important safety information. Belsomra is a controlled substance (Schedule-IV) because it can be abused or lead to dependence.
Belsomra is made by Merck, Sharpe & Dohme Corp. of Whitehouse Station, N.J.
A panel of experts at the US Food and Drug Administration has recommended Merck & Co’s insomnia drug suvorexant when given in lower dosages but rejected the higher dose that the company was seeking.———read more at
Suvorexant (MK-4305) is a dual orexin receptor antagonist in development by Merck & Co.[1][2][3] Suvorexant works by turning off wakefulness rather than by inducing sleep.[4] It is not currently approved for commercial use, but it has completed three Phase III trials.[5]The recent FDA review showed that the drug is associated with increased somnolence the next day and users of higher doses had an increased rate of suicidal ideation. [6] It is one of two such compounds currently in development, the other being GlaxoSmithKline‘s SB-649,868.
Ref:Org.Process Res.Dev-2011-15-367.
PAPER
Mangion IK, * Sherry BD, Yin J, Fleitz FJ. Merck & Co., Rahway, USA
Enantioselective Synthesis of a Dual Orexin Receptor Antagonist.Org. Lett. 2012; 14: 3458-3461
OREXINS A AND B ARE EXCITATORY NEUROPEPTIDES THAT STIMULATE WAKEFULNESS. SUVOREXANT IS A DUAL OREXIN RECEPTOR ANTAGONIST THAT IS IN PHASE III CLINICAL TRIALS FOR THE TREATMENT OF INSOMNIA. THE KEY STEP IN THE ASYMMETRIC SYNTHESIS DEPICTED IS A TANDEM ENZYMATIC TRANSAMINATION–ANNULATION SEQUENCE (F → G → H).
A previous synthesis of suvorexant (N. A. Strotman et al. J. Am. Chem. Soc. 2011, 133, 8362) involved an asymmetric Ru-catalyzed reductive amination in the construction of the diazepane ring. The present route benefits from the circumvention of transition-metal catalysis and dichloromethane as solvent.
To a solution of 22.3 g (78 mmol) of the hydrochloride salt of F-1, 15.9 g (78 mmol) A-2, 12.8 g (94 mmol) 1-hydroxy-7-azabenzotriazole, and 43.1 mL (392 mmol) N-methylmorpholine in 300 mL of DMF was added 22.5 g (118 mmol) EDC and the reaction was stirred overnight at room temperature. The reaction was partitioned between EtOAc and saturated aqueous NaHCO3, washed with water, brine, dried over MgSO4, and concentrated by rotary evaporation. The residue was purified by column chromatography on silica gel (EtOAc/hexanes) to provide G-1 as a colorless gum. Data for G-1: LC/MS: rt=2.22 min; m/z (M+H)=434.2 found; 434.2 required.
A round bottom flask containing a solution of 29.6 g (68.3 mmol) G-1 in 300 mL EtOAc and 200 ml MeOH was evacuated under reduced pressure and purged three times with an atmosphere of N2. To the flask was then added 2.4 g of 20% Pd(OH)2on carbon. The flask was again evacuated under reduced pressure and purged three times with an atmosphere of N2, and then three times with H2. The reaction was stirred under an atmosphere of H2 for three days, then filtered through a pad of celite, rinsing with EtOAc followed by MeOH. The filtrate was concentrated to provide G-2 as a white foam. Data for G-2: LC/MS: rt=0.96 & 1.13 min (see two conformers under these conditions); m/z (M+H)=300.0 found; 300.2 required.
To 21.0 g (70.1 mmol) G-2 in 250 mL DMF was added 29.3 mL (210 mmol) triethylamine and 13.2 g (70.1 mmol) D-1 and the mixture was heated in an oil bath at 75° C. for 2 h. After cooling to room temperature, the reaction was diluted with EtOAc, washed with saturated aqueous NaHCO3, water, brine and dried over MgSO4. Following concentration by rotary evaporation, the residue was purified by flash column chromatography (hexanes/EtOAc) to provide a gum. The gum was stirred in a mixture of 150 ml EtOAc and 300 ml hexanes overnight. Filtration provided G-3 as a white solid. Data for G-3: LC/MS: rt=2.29 min; m/z (M+H)=451.1 found; 451.2 required; HRMS (APCI) m/z (M+H) 451.1631 found; 451.1644 required.
(2) Org.Process Res.Dev.2011,15,367 – 375 reported a synthetic route is as follows:
the two lines above has the following disadvantages: starting materials using highly toxic compound methyl vinyl ketone, methyl vinyl ketone to the eyes, skin, mucous membranes and upper respiratory tract irritation strong, easy to operate when used; and finally to preparation suvorexant, the need to chiral separation, is not conducive to industrial production, but low yield.
(3) W02012148553 and J.Am.Chem.Soc.2011,133,8362 – Scheme 8371 report as follows:
The route disadvantages: starting materials using highly toxic compound methyl vinyl ketone, methyl vinyl ketone to the eyes, skin, mucous membranes and upper respiratory tract irritation strong, easy to operate when used; also use a heavy metal catalyst, high cost, and environmentally unfriendly.
(4) Org.Lett, synthetic route Vol.14, N0.13,2012,3458-3461 reported as follows:
The disadvantage of this route: starting materials using highly toxic compound methyl vinyl ketone, methyl vinyl ketone pairs of eyes, skin, mucous membranes and upper respiratory tract irritation strong.; Additional use of biocatalysis, high cost.
(5) Angew.Chem.1nt.Ed.2011,50,11511 – 11515 reported synthetic route is as follows:
The methyl-2- (benzylamino) ethyl ester (20mmol), (R) _3_ ((tert-butoxycarbonyl) amino) butyric acid (21mmol), 1- hydroxybenzotriazole (25mmol), Sodium hydride (24mmol) added to the flask, anhydrous acetone 50ml, was added with stirring 1 (Shu ^ (25 dirty 01), 301:! 411. The reaction was added 10% citric acid solution, extracted with ethyl acetate, 5% Na2CO3 The organic layer was washed with a solution, and saturated brine, MgSO4 dried, filtered and evaporated to dryness, the product obtained from ethyl acetate and petroleum ether (1: 2, volume ratio) was recrystallized to obtain (yield 97%, mp: 107 ° C, [a] 26D =
21.97 (103.76mg / 20ml, MeOH)).
Example 4: [0074] (R) -4- benzyl-7-methyl-1,4-diaza Synthesis heptane-2,5-dione
The 3g (8.2mmol) (R) – methyl _2_ (N- benzyl _3_ ((tert-butoxycarbonyl) amino) butanamide yl) acetate were added to the flask, and dissolved in ethyl acetate was added IOml added 30ml45% of acetate hydrochloride gas, 25 ° C reaction 4h.Evaporated to dryness, and saturated NaHC03 solution, methylene chloride and ethanol (2: 1, volume ratio) was extracted, MgSO4 organic layer was dried and evaporated to dryness to give a pale yellow oil.It was dissolved in 30ml MeOH and dried added 0.487g (9.02mmol) NaOMe, under nitrogen, 10 ° C reaction 4h.Quenched with saturated NH4Cl solution was added 5 ^ Na2CO3 solution, methylene chloride and ethanol (2: 1, volume ratio) was extracted organic layers were combined, MgSO4 dried, rotary evaporated to give a white solid (yield 98.93%, mp: 122_123 ° C , [a] 26D = 33.49 (112.87mg / 20ml, MeOH)).IH NMR (600MHz, DMS0_d6) δ ppm7.77-7.76 (bd, 1H), 7.33-7.25 (m, 5H), 4.59-4.53 (m, 2H), 4.10-4.02 (m, 2H), 3.65-3.62 ( m, 1H), 2.93-2.90 (m, 1H),
2.76-2.72 (m, 1H), 1.14-1.13 (d, 3H); (FIG. 2) MS (ESI) m / z233.10 ([M + H] +) ..
The 3g (8.2mmol) (R) – methyl _2_ (N_ _ _3 benzyl ((tert-butoxycarbonyl) amino) butanamide yl) acetate were added to the flask, dissolved in dichloromethane was added IOml adding 30ml methylene chloride solution containing 10% of CF3COOH of, 25 ° C reaction 4h.Evaporated to dryness and saturated NaHCO3 solution, methylene chloride and ethanol (2: 1, volume ratio) was extracted, MgSO4 organic layer was dried and evaporated to dryness to give a yellow oil.This was dissolved in 50ml of dry toluene, was added 0.156g (6.5mmol) of sodium hydride, 110 ° C reaction 4h.After cooling to room temperature, quenched with saturated NH4Cl solution, 5% Na2CO3 solution is added, methylene chloride and ethanol (2: 1, volume ratio) was extracted organic layers were combined, MgSO4 dried, rotary evaporated to give a white solid 1.83g (yield 90.34 %, mp: 122-123 ° C, [a J26D = 33.49 (112.87mg / 20ml, MeOH)).
The 3g (8.2mmol) (R) – methyl _2_ (N_ _ _3 benzyl ((tert-butoxycarbonyl) amino) butanamide yl) acetate were added to the flask, methanol was added IOml dissolved, 30ml36% methanol solution of hydrochloric acid gas, 25 ° C reaction 4h.Evaporated to dryness, and saturated NaHC03 solution, methylene chloride and ethanol (2: 1, volume ratio) was extracted, MgSO4 organic layer was dried and evaporated to dryness to give a yellow oil.This was dissolved in 50ml of dry toluene, was added 1.7g (12.3mmol) of potassium carbonate, 110 ° C reaction 8h.After cooling to room temperature, quenched with saturated NH4Cl solution, 5% Na2CO3 solution is added, methylene chloride and ethanol (2: 1, volume ratio) was extracted organic layers were combined, MgSO4 dried, rotary evaporated to give a white solid (yield 95.78%, mp: 122_123 ° C, [a] 26D = 33.49 (112.87mg / 20ml, MeOH)).
The 3g (8.2mmol) (R) – methyl _2_ (N_ _ _3 benzyl ((tert-butoxycarbonyl) amino) butanamide yl) acetate were added to the flask, methanol was added IOml dissolved, 30ml of 36% methanol containing hydrochloric acid gas solution, 25 ° C reaction 4h.Evaporated to dryness and saturated NaHCO3 solution, methylene chloride and ethanol (2: 1, volume ratio) was extracted, MgSO4 organic layer was dried and evaporated to dryness to give a yellow oil.Which was dissolved in 30ml of ethyl acetate and dried, was added 0.88g (16.4mmOl) sodium alkoxide, 10 ° C the reaction 6h.Quenched with saturated NH4Cl solution was added 5 ^ Na2CO3 solution, methylene chloride and ethanol (2: 1, volume ratio) was extracted organic layers were combined, MgSO4 dried, rotary evaporated to give a white solid (yield 93%, mp: 122_123 ° C , [a] 26D = 33.49 (112.87mg / 20ml, Me0H)) ο Example 8:
The 3g (8.2mmol) (R) – methyl _2_ (N- benzyl _3_ ((tert-butoxycarbonyl) amino) butanamide yl) acetate were added to the flask, methanol was added IOml dissolved, 30ml hydrochloric acid gas containing 36% methanol solution, 25 ° C reaction 4h.Evaporated to dryness, and saturated NaHC03 solution, methylene chloride and ethanol (2: 1, by volume) to extract, MgS04 organic layer was dried and evaporated to dryness to give a yellow oil.Which was dissolved in 30ml of dry methanol was added 2.07g (20.5mmol) of triethylamine, 60 ° C the reaction 8h.After cooling to room temperature, quenched with saturated NH4Cl solution, 5% Na2CO3 solution is added, methylene chloride and ethanol (2: 1, volume ratio) was extracted organic layers were combined, MgSO4 dried, rotary evaporated to give a white solid (yield 92.68%, mp: 122_123 ° C, [a] 26D = 33.49 (112.87mg / 20ml, MeOH)).
The 3g (8.2mmol) (R) – methyl _2_ (N_ _ _3 benzyl ((tert-butoxycarbonyl) amino) butanamide yl) acetate were added to the flask, methanol was added IOml dissolved, 30ml hydrochloric acid gas containing 36% methanol solution, 25 ° C reaction 4h.Evaporated to dryness, and saturated NaHC03 solution, methylene chloride and ethanol (2: 1, by volume) to extract, MgSO4 organic layer was dried and evaporated to dryness to give a yellow oil.Which was dissolved in 30ml of dry acetonitrile was added 1.38g (12.3mmol) of potassium t-butoxide, 30 ° C the reaction 8h.Quenched with saturated NH4Cl solution was added 5 ^ Na2CO3 solution, methylene chloride and ethanol (2: 1, volume ratio) was extracted organic layers were combined, MgSO4 dried, rotary evaporated to give a white solid (yield 89.86%, mp: 122_123 ° C , [a] 26D = 33.49 (112.87mg / 20ml, MeOH)).
Example 10:
(R) -1- benzyl-5-methyl-1,4-Synthesis diazepan the
A 1.4g (R) -4- benzyl-7-methyl-diaza heptane _2,5_ _1,4_ dione (6mmol) was dissolved in 60ml dry THF, was added portionwise under ice- 1.35g LiAlH4 (36mmol), 25 ° C was stirred for 4h.Cooled to -10 ° C, was added 1.5mlH2O quenched and then 1.5mll5% NaOH, 4.5ml H20, part MgSO4, stirring lh, filtration, spin dried to give 1.2g oil (yield 97.56%, [a] 29D = -5.87 (200.86mg / 20ml, CHCl 3)).ee> 99%, Chrom Techchiral-AGP150 * 4mm Mobile phase: Ammonium dihydrogen sulfate (IM): acetonitrile = 99: 1, column temperature: 30 ° C, flow rate: 0.5ml / Hiin0 IH NMR (600MHz, DMS0_d6) δ ppm7.32-7.20 (m, 5Η), 3.57 (s, 2Η), 3.48 (bs, 1Η), 2.99-2.95 (m, 1Η), 2.86-2.82 (m, 1Η), 2.72-2.68 (m, 1Η ), 2.65-2.61 (m, 1Η), 2.58-2.49 (m, 3Η), 1.75-1.70 (m, 1Η), 1.46-1.41 (m, 1Η), 1.01-1.00 (d, 3Η); (Figure 3 .) MS (ESI) m / z205.10 ([M + H] +) [0095] Example 11:
(R) -1- benzyl-5-methyl-1,4-Synthesis diazepan the
A 1.4g (R) -4- benzyl-7-methyl-diaza heptane _2,5_ _1,4_ dione (6mmol) was dissolved in 60mlTHF TEMPERATURE dropwise 2 equivalents of borane ( 12mm0l), reflux 8h.Cooled to _10 ° C, quenched by addition of methanol, adjusted pH = 3, stirred for 2h, sodium carbonate adjusted to pH = 10, extracted with methylene chloride three times, the combined organic layer, MgSO4 drying, rotary evaporation.(Yield 95.32%, [a] 29D = -5.87 (200.86mg / 20ml, CHCl 3)).[0098] Example 12:
(R) -1- benzyl-5-methyl-1,4-Synthesis diazepan the
The (R) -4- benzyl-7-methyl-1,4-diaza heptane-2,5-dione (5mmol) was dissolved in 15ml dry THF, was added under ice-cooling to a solution of Ig sodium boron (27mmol) in 15ml dry THF hydride was added dropwise a solution of iodine in 20ml THF 12mmol dried under nitrogen, at reflux for 6h.Cooled to (TC, quenched 5ml3N HCl was added, followed by addition of 8ml3NNaOH, liquid separation, the aqueous layer was extracted three times with ether, the combined organic layer was washed with saturated brine, MgSO4 drying, filtration, spin dry (yield 90.34%, [a ] 29D = -5.87 (200.86mg / 20ml, CHC13)).
Example 13:
(R) – (4_-Benzyl-7-methyl-1,4-diazepan-1-yl) (5-methyl _2_ (2H-1,2,3_ three
Synthesis of 2-yl) phenyl) methyl ketone
The 3g (R) -1- benzyl-5-methyl-1,4-diazepane (14.7mmol), 3.66g5_ methyl -2- (2Η-1, 2,3- triazol-2-yl) benzoic acid (18.03mmol) was dissolved in DMF, 2.43gHOBt (18.55mmol), 6ml TEA (42.75mmol), 3.45g EDC (17.99mmol), warmed to 50 ° C, the reaction 2h.Was added a saturated NaHCO3 solution and EA, the aqueous layer was washed three times with EA, the combined organic layers.The organic layer was washed with citric acid solution, the product salified fully into the aqueous phase, the aqueous phase was washed with EA after the addition of sodium carbonate to adjust the pH> 9, EA and washed three times, the organic layers combined, washed with water and saturated brine, MgSO4 dried, rotary dried, PE and EA (4: 1) and recrystallized (yield 98.36%, mp: 108-109 ° C, [α] 31D = -58.37 (202.5mg / 20ml, MeOH)).IH NMR (600MHz, DMS0_d6) δ ppm8.00-7.76 (m, 3H), 7.37-7.17 (m, 7H), 4.40-4.09 (m, 1H), 3.63-3.48 (m, 2H), 3.44-3.02 ( m, 3H), 2.82-2.75 (m, 1H), 2.63-2.47 (m, 1H), 2.63-2.14 (m, 5H), 2.02-1.63 (m, 2H), 1.17-0.99 (m, 3H); (Figure 4) MS (ESI) m / z390.30 ([M + H] +) [0105] Example 14:
The 3g (R) -1- benzyl-5-methyl-1,4-diazepane (14.7mmol), 2.98g5_ methyl -2- (2H-1, 2,3- triazol-2-yl) benzoic acid (14.7mmol) was dissolved in methylene chloride, was added 18.55mmolHOAt, 6ml TEA (42.75mmol), 2.86g CDI (17.64mmol), 30 ° C reaction 4h.Was added a saturated NaHC03 solution and EA, the aqueous layer was washed three times with EA, the combined organic layers.The organic layer was washed with citric acid solution, the product salified fully into the aqueous phase, the aqueous phase was washed with EA after the addition of sodium carbonate to adjust the pH> 9, EA and washed three times, the combined organic layer was washed with saturated brine paint, MgS04 drying, spin dry, PE and EA (4: 1) and recrystallized (yield 96.45%, mp = 108-109 ° C, [a J31D = -58.37 (202.5mg / 20ml, MeOH)).
Example 15:
(R) – (4_-Benzyl-7-methyl-1,4-diazepan-1-yl) (5-methyl _2_ (2H-1,2,3_ triazol – 2- yl) phenyl) -methanone [0110] The 3g (R) -1- benzyl-5-methyl-1,4-diazepane (14.7mmol), 3.28g5_ methyl – 2- (2Η-1,2,3- triazol-2-yl) benzoic acid (16.17mmol) was dissolved in acetone was added 2.43gHOBt (18.55mmol), 6ml TEA (42.75mmol), 3.33gDCC (16.17mmol) After the addition of sodium carbonate, 3 (TC reaction 4h. Saturated NaHCO3 solution was added and EA, the aqueous layer was washed three times with EA, the combined organic layers. The organic layer was washed with citric acid solution, the product salified fully into the aqueous phase, the aqueous phase was washed with EA adjust pH> 9, EA and washed three times, the organic layers combined, washed with water and saturated brine, MgSO4 dried, rotary dried, PE and EA (4: 1) and recrystallized (yield 92.43%, m.ρ .: 108-109 .. , [a J31D = -58.37 (202.5mg / 20ml, MeOH)).
A 2.08g (R) – (4_ _1,4_ Benzyl-7-methyl-diazepan-1-yl) (5_-methyl -2- (2Η-1, 2, 3- triazol-2-yl) phenyl) methyl ketone (7.2mmol) was dissolved in 20ml THF, 10% of the PdC12,50 ° C through the H2 reaction 2h.Filtration, rotary evaporation to give the product (yield 93.24%, [a] 26D = -14.36 (199.12mg / 20ml, MeOH)).
A 2.08g (R) – (4_ _1,4_ Benzyl-7-methyl-diazepan-1-yl) (5_-methyl -2- (2H-1, 2, 3- triazol-2-yl) phenyl) methanone (7.2mmol) was dissolved in 20ml of methanol was added 10% Pd / C, was added ammonium formate (21.6_ο1), the reaction was refluxed for 6h.Filtration, rotary evaporation to give the product (yield 92.68%, [a] 26D = -14.36 (199.12mg / 20ml, MeOH)).
Example 19
Synthesis Suvorexant of
To 0.9g (R) – (7- methyl-1,4-diazepan-1-yl) (methyl 5_ _2_ (2H-1,2,3_ triazol-2 yl) phenyl) methanone (3.0lmmol) of IOml DMF was added 0.57g2, 5- dichlorobenzene and oxazole (3.03mmol), 0.91g TEA (9mmol), heated to 75 ° C, the reaction 2h.Cooled to room temperature, EA dispersion, washed with a saturated NaHCO3 solution, saturated brine, MgSO4 dried, rotary evaporated to give a white solid (yield 93.02%, mp: 128-129 ° C, [a] 3C1.9D = -11.7 (199.99 mg / 20ml, MeOH)).IH NMR (600MHz, DMS0_d6) δ ρρm8.05-7.88 (m, 2Η), 7.82-7.78 (m, 1Η), 7.42-7.25 (m, 2Η), ζ, 06-7.00 (m, IH), 4.29- 4.06 (m, 1Η), 4.01-3.72 (m, 2Η), 3.66-3.49 (m, 2Η), 2.10 (s, 3Η), 2.06-2.01 (m, IH), 1.50 (m, 1Η), 1.78- 1.50 (m, 1Η), 1.14-1.13 (d, 3Η); (FIG. 6) MS (ESI) m / z451.20 ([Μ + Η] +).
The compound of the formula I is disclosed as an antagonist of orexin receptors in US Patent 7,951,797, US Patent Application Publication US 2008/0132490, PCT PatentPublication WO 2008/069997, Cox et al, J. Med. Chem. 2010, 53, 5320-5332, Strotman et al, JACS, 2011, 133(21), 8362-8371, and Baxter et al, Org. Process Res. & Dev., 201 1, 15(2) 367- 375.
This compound is disclosed as having activity in antagonizing the human orexin-1 (OX1) receptor with a Ki of 0.55 nM and in antagonizing the human orexin-2 (0X2) receptor with a Ki of 0.35 nM. The processes disclosed in US Patent 7,951,797, US Patent Application Publication US 2008/0132490, PCT Patent Publication WO 2008/069997, Cox et al, J. Med. Chem. 2010, 53, 5320-5332, Strotman et al, JACS, 201 1, 133(21), 8362-8371, and Baxter et al, Org. Process Res. & Dev., 2011, 15(2) 367-375 are lengthy, suffer from low yields, necessitate multiple protecting groups, rely on chiral chromatography to prepare a single isomer and require microwave technology to prepare the acid intermediate. Relative to the processes disclosed in US Patent 7,951,797, US Patent Application Publication US 2008/0132490, PCT Patent Publication WO 2008/069997, Cox et al, J. Med. Chem. 2010, 53, 5320-5332, Strotman et al, JACS, 2011, 133(21), 8362-8371, and Baxter et al, Org. Process Res. & Dev., 201 1, 15(2) 367- 375, the present invention may provide improved processes for the efficient, scalable, chromatography-free and cost-effective preparation of the formula I, to give higher isolated yield of the subject compound.
EXAMPLE 1
2. DMF
5-Chloro-l,3-benzoxazole-2-thiol (9a)
2-Amino-4-chlorophenol (2.50 kg, 17.4 mol) was charged to a vessel and suspended in water (52 L) and methanol (10.4 L). High dilution was required to prevent slow and difficult filtration of the product. The mixture was stirred, cooled to 0 °C, then thiophosgene (2.00 kg, 17.4 mol) was added to the suspension ensuring that the internal temperature remained at 5 °C throughout the addition. Water (8 L) and methanol (2 L) were added to aid stirring and the slurry was warmed to 13 °C for 1 h, followed by aging at 20 °C for a further 1 h. The slurry was then filtered and the solid washed with water (5 L). The batch was repeated and combined to dry in a vacuum oven (T = 40 °C) for 15 h to give 9-a (5.81 kg, 31.3 mol). The data corresponds to the commercially available material. XH NMR (400 MHz, d6-DMSO): δ 7.51 (d, 1 H, J = 9.2 Hz), 7.307.26 (m, 2 H). 13C NMR (100.6 MHz, d6-DMSO): δ 181.2, 147.4, 133.1, 129.7, 123.9, 1 11.6, 110.8. HRMS (ESI): m/z [M+ + H] calcd for C7H4CINOS: 185.9780; found: 185.9785.
Thiol 9a (10.5 kg, 54.6 mol) was added to a vessel and suspended in DCM (141 kg). Oxalyl chloride (10.4 kg, 82.3 mol) was added (slightly endothermic) followed by DMF (40.0 kg, 547 mol) over 1.25 h, such that the batch temperature was≤ 25 °C. The batch was aged at 20 °C for approximately 30 min, HPLC analysis showed reaction to be complete. The batch was cooled to 10 °C then triethylamine (16.64 kg, 164.4 mol) was added via a sub-surface sample line at such a rate as to maintain a batch temperature of≤ 10 °C. A sub-surface addition protocol was required to prevent build up of triethylamine hydrochloride solid on the walls of the vessel. The batch was cooled to 0 °C, then a solution of N-Boc-ethylenediamine (10.5 kg, 61.2 mol) in DCM (10 kg) was added such that the batch temperature was≤ 10 °C. The reaction was warmed to 20 °C and stirred for 2.5 h, HPLC analysis showed the reaction to be complete. Water (63.6 kg) was charged to the batch and the mixture stirred for 5 min. The layers were separated and the aqueous phase re-extracted with DCM (42.2 kg). The organic solutions were then combined and approximately half of the total DCM volume was distilled from the batch under vacuum whilst maintaining a temperature of≤ 40 °C. MeCN (83.3 kg) was then added and the remaining DCM removed by distillation (0.5 mol % DCM left by XH NMR wrt MeCN). MVK (4.61 kg, 65.8 mol) was added to the batch followed by DBU (4.17 kg, 27.4 mol) such that the temperature was≤ 20 °C. The batch was aged for 10 h at 20 °C then analyzed by HPLC. The reaction was then diluted with water (42.4 kg) and aged for a further 30 min. The mixture was filtered and the slurry washed with MeCN (33.3 kg). The solid was washed with MeCN (-10 L) then dried in a vacuum oven (T = 60 °C) for 22 h. MVK adduct 10 (15.5 kg) was isolated as an off-white solid, mp 145-148 °C. ¾ NMR (400 MHz, CDC13): δ 7.24 (d, 1 H, J = 2.3 Hz), 7.09 (d, 1 H, J = 8.5 Hz), 6.91 (dd, 1 H, J = 8.5, 2.3 Hz), 5.06 (s, 1 H, br), 3.73 (t, 2 H, J = 6.7 Hz), 3.63 (t, 2 H, J = 6.1 Hz), 3.37 (d, 2 H, br), 2.89 (t, 2 H, J = 6.7 Hz), 2.14 (s, 3H), 1.33 (s, 9 H). 13C NMR (100.6 MHz, CDC13): 8 206.7, 163.0, 156.0, 147.4, 144.6, 129.2, 120.3, 116.6, 109.2, 79.4, 49.3, 44.3, 41.9, 39.1, 30.2, 28.3. HRMS (ESI): m/z [M+ + H] calcd for
382.1534; found: 382.1544.
EXAMPLE 2
□ HMDS, THF/hexane (3.6:1.0), -25 to -15 °C; NBS
5-Chlorobenzoxazole (3-2)
To a 250 mL 3-neck round bottom flask equipped with a distillation head, glass stopper, septum, thermocouple and magnetic stir bar was charged 2-amino-4-chlorophenol (20.00 g, 0.139 mol). The solid was dissolved in THF (60 mL) and p-TsOH (0.265 g, 1.39 mmol) was added. The brown solution was warmed to 60 °C over 10 min and aged for 90 min. HPLC assay of the reaction mixture showed 1 LCAP unreacted starting material. The temperature was increased from 60 °C to 74 °C, and at 63 °C solvent distillation began. A total of 58 mL was collected during the first distillation. The mixture was diluted with THF (60 mL) and a total of 67 mL of solvent was removed between 71 and 84 °C. The mixture was again diluted with THF (60 mL) and 61 mL of solvent was removed between 74 and 1 14 °C. The dark brown solution was cooled to room temperature. The final mass of the solution was 27.96 g. Analysis of the crude stream by XH NMR showed 0.1 wt% MeOH present in the sample. XH NMR (500 MHz, CDC13): δ = 8.10 (s, 1H), 7.76 (d, J= 1.5 Hz, 1H), 7.50 (d, J= 8.7 Hz, 1H), 7.36 ppm (dd, J= 8.7, 1.7 Hz, 1H).
A 500 mL 3-neck round bottom flask equipped with a septum, thermocouple, 125 mL addition funnel, inert gas inlet and magnetic stir bar was purged with nitrogen for 10 min. Hexamethyldisilazane (42 mL, 0.20 mol) and THF (78 mL) were charged against positive nitrogen pressure. The addition funnel was charged with a hexane solution of n-butyllithium (78.0 mL, 195 mmol). The amine solution was cooled to -52 °C and n-butyllithium was added over 84 min, resulting in a temperature increase to 12.5 °C over the course of the addition. The resulting lithium hexamethyldisilazide solution was removed from the cooling bath and aged for 30 minutes. To a 500 mL 3 -neck round bottom flask equipped with a septum, thermocouple, inert gas inlet and magnetic stir bar was charged 5-chlorobenzoxazole (20.00 g, 130 mmol). The gray solid was dissolved in THF (100 mL) and the resulting colorless solution was cooled to -25 °C. The freshly prepared lithium hexamethyldisilazide solution was added via cannula over 80 minutes. The temperature of the anion solution was maintained between -25 and -15 °C during the addition. The resulting dark brown solution was aged for 90 minutes between -25 and -15 °C. To a 1000 mL 3-neck round bottom flask equipped with a Claisen adapter, septum,
thermocouple, inert gas inlet, stir rod bearing, and blade was charged THF (100 mL) and N- bromosuccinimide (34.8 g, 195 mmol). The resulting slurry was cooled to -20 °C and the anion solution was added via cannula over 150 minutes. During the addition the anion solution and reaction mixture were maintained between -25 and -15 °C. The resulting brown slurry was removed from the cooling bath and aged for 50 minutes while warming to room temperature. To the resulting bromide slurry was added a solution of ethanolamine (12.6 mL, 208 mmol) in MeCN (38 mL) via syringe pump over 5 hours. During the addition the reaction temperature was maintained between 20 and 27 °C. The resulting brown slurry was aged at room temperature overnight. The reaction mixture was cooled in an ice water bath and the septum replaced with a 50 mL addition funnel charged with concentrated HC1 (32 mL, 390 mmol). The acid solution was added over 10 min, during which time the addition the temperature increased from 10 to 20 °C. The reaction mixture was removed from the ice water bath and aged for 5 min. A 20% (w/w) solution of K2HPO4 in water (170 mL) was added and the resulting biphasic mixture was transferred to a seperatory funnel. The flask was washed with THF (3x, 10 mL) and the washings were added. The aqueous phase was cut; the organic phase was washed with 20% (w/w) K2HPO4 in water (200 mL), separated and analyzed. The crude reaction stream had a total mass of 396.47 g. By quantitative HPLC assayed 25.81 g of 3-3 in the organic phase. XH NMR (500 MHz, DMSO-i¾): δ = 8.17 (t, J= 5.6 Hz, 1H), 7.34 (d, J= 8.4 Hz, 1H), 7.25 (d, J= 1.8 Hz, 1H), 6.97 (dd, J= 8.4, 1.8 Hz, 1H), 4.81 (t, J= 5.4 Hz, 1H), 3.56 (q, J= 5.7 Hz, 2H), 3.35 pm (q, J= 5.8 Hz, 2H).
To a 1000 mL 3-neck round bottom flask equipped with a septum, thermocouple, inert gas inlet and magnetic stir bar was charged 3-3 (25.2 g, 119 mmol). To this flask was added 126 mL DMF, 12.2 mL methyl vinyl ketone (148 mmol) and 0.119 mL 10M NaOH (1.19 mmol). The reaction was then aged for 6 hours, at which time conversion was judged to be complete by HPLC. The solution was diluted with 252 mL iPAc and cooled to 0 °C, then 23.1 mL Et3 (166 mmol) followed by dropwise addition of 12.0 mL methanesulfonyl chloride (154 mmol) over 45 minutes, maintaining internal temperature less than 10 °C. After a further 30 minutes, conversion was judged to be complete by HPLC. The solution was washed with 3x 63 mL 5 w/w% aqueous aHC03 solution, then 66 mL water. After cutting the aqueous layer, the organics were reduced to approximately two volumes or 50 mL iPAc. The organics were then agitated by an overhead stirrer during slow addition of 151 mL n-Heptane over 4 hours. Over this time a crystalline white precipitate developed, and was allowed to stir overnight. At this time there was a thick slurry, which was filtered and washed with 2x 50 mL 90: 10 n- HeptaneTPAc, and after drying with a nitrogen stream over the filter pad, 3-4 was obtained as a white crystalline solid (34.6 g., 96 mmol). ‘H NMR (500 MHz, CDC13): δ = 7.29 (s, 1H), 7.16 (d, J= 8.2 Hz, 1H), 6.97 (d, J= 7.8 Hz, 1H), 4.46 (s, 2H), 3.92 (s, 2H), 3.81 (t, J= 5.9 Hz, 2H), 2.98-2.92 (m, 5H), 2.16 (s, 3H).
EXAMPLE 3
5-Chloro-2-((R)-5-methyl-[l,4]diazepan-l-yl)-benzooxazole hydrochloride (R-11) To a 1000 mL 3 -necked flask was charged isopropylamine hydrochloride (25.8 g., 270 mmol) and 525 mL 0.1 M aqueous triethanolamine solution. To this was added 750 mg pyridoxal 5′-phosphate hydrate (PLP) and 3.0 g of the transaminase polypeptide having the amino acid sequence SEQ ID NO: l and the suspension was stirred until all components dissolved. The transaminase polypeptide having the amino acid sequence SEQ ID NO: 1 was obtained as disclosed in US Patent Publication US 2010/0285541 for the identical sequence “SEQ ID NO: 1 10” therein. The solution was heated to 40 °C and the pH of the solution was adjusted to pH 8.5 with an aqueous 4M solution of isopropylamine. Mesylate 3-4 was added as a 225 mL DMSO solution via syringe over 6 hours, and the resulting mixture stirred for a further 5 hours. At this time, the solution was poured into a 3L separatory funnel and extracted with 1.5 L of 1 : 1 iPAc:IPA. The aqueous layer was cut then extracted again with 750 mL 4: 1 iPAc:IPA. The organics were combined, then washed with 750 mL brine. Then the organics were concentrated with IPA flushing to establish a 45 mL solution in IPA which was then treated with 4.6M HC1 in IPA (9.94 mL, 45.7 mmol) via dropwise addition. The resulting solution was stirred vigorously while 52 mL IP Ac was added slowly over 5 hours, creating a slurry of HQ salt 6. The slurry was then slowly cooled to 0 °C and allowed to stir overnight. At this time the slurry was filtered and dried with a nitrogen stream over the filter pad, providing R-11 as a white crystalline solid (7.80 g., 25.8 mmol). ¾ NMR (500 MHz, CD3OD): δ = 7.13-7.10 (m, 2H), 6.97 (dd, J= 8.2, 1.8 Hz, 1H), 3.99-3.79 (m, 3H), 3.67-3.57 (m, 3H), 3.39-3.33 (m, 1H), 2.24 (s,
1H), 2.12-2.07 (m, 1H), 1.42 (d, J= 6.7 Hz, 3H).
EXAMPLE 4
19 5
5-Methyl-2-[l,2,3]triazol-2-yl-benzoic acid (5) The iodide 19 (6.04 kg, 23.0 mol), THF (45 L) and DMF (9.0 L) were charged to a vessel. Copper iodide (218 g, 1.15 mol) and potassium carbonate (7.94 kg, 57.4 mol) were added and the mixture heated to an internal temperature of 40 °C. 1,2,3-Triazole (3.16 kg, 46.0 mol) was added as a solution in THF (6.0 L) over half an hour (no exotherm) and heating continued to 65 °C (again no exotherm observed) and the reaction monitored by HPLC. Once complete N,N-dimethylethylenediamine (244 mL, 2.30 mol) was added and mixture cooled to RT. Aqueous 3.6 M HC1 (36 L) was added (exotherm) and the mixture extracted twice with ethyl acetate (2 x 30 L). The combined organics were washed with LiCl solution (2 x 20 L). The acid solution assayed for 3.79 kg of 5 (81%) and 4.64 kg of 5 and 20 combined (99%). A solution of acids 5 and 20 (approx. 4.64 kg, 22.9 mol) in THF and EtOAc (approx. 1 10 L) was concentrated to low volume. THF (90 L) was added and the solvent composition checked by XH NMR to ensure most ethyl acetate had been removed. Sodium tert-butoxide (2.42 kg, 25.2 mol) was added slowly as a solid over 1-2 h (slight exotherm), allowing the sodium salt to form and stirred overnight at RT. The liquors showed a 45:55 ratio of product: starting material and the solid was collected by filtration, washed with THF (2 x 20 L) and dried in a vacuum oven (T = 40 °C) for 15 h to afford 4.22 kg of crude sodium salt. The crude sodium salt (4.22 kg, 14.9 mol) was charged to a 50 L vessel and 3.6 M HC1 (21.2 L) was added with cooling. The slurry was then stirred at room temperature for 16 h and the off-white solid isolated by filtration. The cake was washed with water (11 L) and iP Ac/Heptane (2 x 5L), then dried in a vacuum oven (T = 35 °C) for 15 h to give 3.10 kg of crude acid 5 (97.9 LCAP, 92 wt%, corrected weight 2.85 kg, 61% yield from 19). The acid 5 (2.85 kg corrected, 14.0 mol) was charged to a 50 L vessel and EtOAc (28 L) and dilute 0.22 M HC1 (14 L) were added and the mixture stirred until two clear phases resulted. The aqueous layer was removed and the organic layer filtered to remove any particulate matter. The ethyl acetate was reduced to about 8 L and then heptane (15.6 L) was added over 1 h and the liquors sampled to check for appropriate losses. The solid was isolated by filtration, washed with heptane:ethyl acetate (3 : 1 , 4 L) and dried on the filter under nitrogen to give 2.81 kg of acid 5. m.p. 167.5 °C. XH NMR (400 MHz, d6-DMSO): δ 12.09 (br s, 1H), 8.04 (s, 1H), 7.62 (d, 1H, J = 8.4 Hz), 7.58 (d, 1H, J = 1.2 Hz), 7.49 (dd, 1H, J = 8.4, 1.2 Hz), 2.41 (s, 3H). 13C NMR (100.6 MHz, d6-DMSO): δ 168.0, 139.2, 136.4, 135.8, 132.5, 130.3, 128.7, 124.8, 20.9. HRMS (ESI): m/z [M+ + H] calcd for C10H9N3O2: 204.0773; found: 204.0781. EXAMPLE 5
A round bottom flask was charged 6.86 g of 5-methyl-2-[l,2,3]triazol-2-yl- benzoic acid (5) along with 7.0 vol or 70 mis of dry iPAc (KF < 200 ppm) forming a slurry. To this was charged 0.73 g of DMF then the system was purged thoroughly with nitrogen and temperature was set at 20°C-25°C. 5.04 g of oxalyl chloride was added while maintaining 20°C- 25°C and controlling off-gassing since it is extremely vigorous. With the feed of oxalyl chloride the previous slurry dissolved. The batch was aged for 1 hr, sampled for acid chloride formation (< 1 LCAP) and allowed to proceed to amidation. In a separate vessel a solution of potassium carbonate was prepared in 5.0 vol or 50 mL water (note: exotherm). The solution was cooled to 0 °C. When acid chloride (above) was prepared, added 2.5 vol or 25 mL iPAc to the aqueous solution with overhead stirring, then added 10.0 g. amine hydrochloride salt (R-ll) to solution, and stirred for 15 minutes. Then using a cannula, the acid chloride solution was transferred over from separate vessel over the course of 1 hour, maintaining less than 5°C internal temperature. The vessel was flushed with 2.5 vol or 25 mL iPAc and sampled to determine completion. The slurry was heated to 40 °C. Upon reaching 40 °C, 1.5 vol or 15 mL Acetonitrile was and agitated for 5 minutes, and all material went into solution (98% AY observed). Agitation was stopped. After phase separation, the aqueous layer was cut, the organics were stirred with DARCO (10 wt% 6 basis) at 40°C for 3 hours, then filtered hot and taken through to
crystallization. Additional product was recovered from the carbon with an iPAc flush.
The batch was concentrated in iPAc and flushed to 7.5 vol (L/Kg of 1) and heated to 80-85C until complete dissolution. The solution was cooled to 65 °C linearly over 2 hrs, and the agitation speed was adjusted to high. At 65 °C, the solution was charged with 0.3 wt% seed in n-Heptane and aged for 1 hour. After the age and confirmation of the seed bed, the batch was cooled to 45 °C over 2.5 hrs. At this time a solvent switch was conducted at constant volume to a ratio of 90: 10 n-Heptane: iP Ac. The material was filtered hot at 45 °C, the cake was washed with 3 vol (L/Kg of 1) of 90: 10 n-Heptane :iP Ac twice, followed by 3 vol (L/Kg of 1) of n- Heptane twice. The cake was dried at 70 °C under vacuum to give 14.4 g. 1 (31.8 mmol,) as a crystalline white powder.
A reaction vessel was charged with 213.4 g of triazole acid (5) along with 7.4 vol or 2236 mis of dry iPAc (KF < 200 ppm) forming a slurry. To this charge was added 21.93 g of DMF then the system was purged thoroughly with nitrogen and temperature was maintained at 20- 25C. Charged 152.3 g of oxalyl chloride while maintaining 20-25C and control of off-gassing since it is extremely vigorous. With the feed of oxalyl chloride the previous slurry all dissolved. The batch was aged for 1 hr. The reaction was sampled for Acid Chloride formation (< 1 LCAP) and proceeded to distillation. Distillation was conducted down to 11 18 ml or constant volume distillation using 7.4 vol of fresh iPAc under vacuum maintaining less than 30°C.
In a separate vessel prepared a solution of 302.2 g of amine hydrochloride salt (R-ll) in 15.3 vol or 4624 mis of dry iPAc (KF < 200 ppm) to form a slurry. Then transferred the acid chloride solution using a cannula over from a separate vessel followed by flushing the vessel with 6.9 vol or 2085 mis of iPAc. With the amine and acid chloride in the same vessel began addition of 404.8 g of triethylamine. This charge was made over 1 to 4 hrs at a temperature between 20-40C with a desired control of the temperature between 20-30C. Once feed of the TEA was complete, the batch was aged for lhr and then sampled to determine completion.
Once the batch was complete, charged 7.4 vol of water or 2236 mis and then heated the solution to 40C. Once at 40C, the mixture was aged 5 minutes then agitation was stopped. The phases separated but there was an appreciable rag layer so it was allowed to settle and the rag was cut along with the aqueous layer. The aqueous rag was filtered then the aqueous layer was back extracted with 3.5 vol or 1058 ml of iPAc and all iPAc layers were combined.
The batch was recycled in iPAc (~60 g per kg of iPAc) via a Cuno filter (1 bundle per 39 Kg Amine HC1 Salt) for several hours at 40°C. The batch was drummed off through a sparkler filter and additional material was recovered from the carbon with an iPAc flush.
The batch was concentrated in iPAc and flushed to 7.5 vol (L/Kg of product) and heated to 80-85°C until complete dissolution. The mixture was cooled to 65°C linearly over 2 hrs, and agitation speed was adjusted to high from this point forward. At 65°C, the mixture was charged with 0.3 wt% of [(R)-4-(5-chloro-benzooxazol-2-yl)-7-methyl-[l,4]diazepan-l-yl]-(5- methyl-2-[l,2,3]triazol-2-yl-phenyl)-methanone seed in n-Heptane and aged for 1-3 hour. After the age and confirmation of the seed bed, the batch was cooled to 45°C over 2.5 hrs. A solvent switch was conducted at constant volume to a ratio of 90: 10 n-Heptane :iP Ac.
The batch was wet milled to a uniform particle size and filter hot at 45C. The cake was washed with 3 vol (L/Kg of product) of 90: 10 n-Heptane :iP Ac twice, followed by 3 vol (L/Kg of product) of n-heptane twice. The cake was dried at 70°C under vacuum.
Suvorexant (MK-4305) is a potent dual Orexin antagonist under development for the treatment of sleep disorders at Merck. The key transformation is an asymmetric Ru-catalyzed transfer hydrogenation (using a modified Noyori RuCl(p-cymene)(DPEN) complex) of an in-situ generated cyclic imine resulting in the formation of the desired chiral diazepane in 97% yield and 94.5% ee. Mechanistic studies have revealed that CO2 (derived from the formic acid) has pronounced effect on reaction outcome. Studies have determined that the efficiency of the Ru-catalyst, the composition of the resulting amine (via carbamate formation), and the reaction kinetics are mediated by the amount of CO2 generated during the reaction. The efficiency of the reductive-amination can be enhanced by either purging the CO2 or by trapping the newly formed nucleophilic secondary amine.
A new synthetic route to drug candidate 1, a potent and selective dual orexin antagonist for the treatment of sleep disorders, has been developed. The key acyclic precursor 10 was prepared in a one-step process in 75% isolated yield from commercially available starting materials using novel chemistry to synthesize 2-substituted benzoxazoles. A reductive amination was followed by a classical resolution to afford chiral diazepane (R)-11. Finally, coupling of (R)-11 with acid 5 furnished the desired drug candidate 1.
The amine DBT salt 16 (5.67 kg, 9.09 mol) was charged to a vessel and inerted. DCM (28 L) was added, followed by 4 N sodium hydroxide solution (prepared from 10 N NaOH [22.4 L] and water [36 L]). The slurry was then stirred at ambient temperature for 1 h until a solution was obtained. The layers were separated, and the aqueous phase was treated with sodium chloride solution (10.1 kg in 20 L water). DCM (5 L) was then added and the biphasic mixture stirred for 10 min before separating the layers. The combined organic layers were then concentrated under reduced pressure to a 10 L volume. The solution of the free amine was used directly in the next reaction
The triazole acid 5 (13.25 kg, 65.2 mol), DCM (88 L), and DMF (1.35 L, 17.4 mol) were charged to a vessel, and the resulting suspension was cooled to 0 °C. Oxalyl chloride (8.28 kg, 65.2 mol) was added portionwise, keeping the internal temperature between 5 and 10 °C (the anhydride formed above 10 °C), and then the reaction was aged for 30 min at this temperature. HPLC analysis showed acid 5 remained; an additional charge of oxalyl chloride (160 g, 1.26 mol) was made, and the solution stirred at 5 °C for 30 min. A solution of the amine (R)-11 (16.5 kg, 62.1 mol) and triethylamine (13.19 kg, 130.0 mol) in DCM (∼8 L) was added to the acid chloride over 30 min, keeping the internal temperature less than 15 °C. The resulting slurry was aged for 30 min and then quenched by the addition of water (167 L) over 10 min, keeping the internal temperature <15 °C. The lower organic layer was removed and then concentrated under atmospheric pressure to a volume of 100 L. Assay at this stage showed 27.3 kg 1, 98%. The solution was solvent switched to MeCN (∼560 L, 20 mL/g) by distillation under reduced pressure at <50 °C. The MeCN solution was treated with Ecosorb C-941 (2.8 kg) slurried in MeCN (10 L). The resulting slurry was aged for 30 min and then filtered through a Solka Flok pad and a 0.1 um cartridge filter, washing with MeCN (2 × 30 L). The MeCN filtrate was concentrated under reduced pressure at <50 °C to a final volume of ∼112 L. The slurry was cooled to 25 °C and water (280 L) added over 40 min. The resulting slurry was aged at 20 °C for 1 h and then filtered, washing the cake with 5:1 water/MeCN (60 L) followed by water (40 L). The solid was dried in the vacuum oven with nitrogen purge overnight at 50 °C. The final target 1 was isolated as a white solid, 26.72 kg, 95%, 98.5% ee, 99.6 LCAP, mp 153.1 °C.
The 1H NMR data for this compound was extremely complicated due to its existence as four rotamers. These rotamers did not coalesce during high-temperature experiments.(4)
[α]25D −11.8 (c 1.0, MeOH) for a sample of 97.8% ee. HRMS (ESI): m/z [M+ + H] calcd for C23H23ClN6O2: 451.1649; found: 451.1640.
A highly regioselective halogenation of 2-substituted-1,2,3-triazoles was developed via sp2 C–H activation. This method is compatible with halogen atoms, as well as electron-donating and electron-withdrawing groups. Meanwhile, the strategy is also efficient for the synthesis of a key intermediate of Suvorexant.
PAPER
2.1. Synthesis of (R)-methyl 2-(N-benzyl-3-((tert-butoxycarbonyl)amino)butanamido)acetate (3)
To a solution of methyl 2-(benzylamino)acetate (compound 10, 50.14 g,0.28 mol),(R)-3-((tert-butoxycarbonyl)amino)butanoic acid (50.75 g,0.25 mol),1-hydroxy-1H-benzotriazole (41.88 g, 0.31 mol),and dry triethylamine (37.95 g,0.38 mol) in 320 mL of DMF was added EDC hydrochloride (57.51 g,0.30 mol),and the reaction was stirred for 5 h at room temperature. The reaction was partitioned between EtOAc and 10% aqueous citric acid,the layers were separated and the organic was washed with 5% aqueous Na2CO3,then with brine,dried over MgSO4 and concentrated by rotary evaporation. The residue was recrystallized from a mixture solvent (PE:EtOAc = 2:1) to provide compound 3 as a white solid, 83.01 g in 91% yield. Mp: 107 ℃,[α]D 25 22.0 (c0.52,MeOH). 1H NMR (600 MHz,DMSO-d6): δ 7.38-7.23 (m,5H),6.73-6.72 (d,1H, J = 6 Hz),4.75-4.43 (m,2H),4.31-3.95 (m,2H),3.89-3.87 (t,1H, J = 12 Hz),3.64-3.62 (d,3H,J = 12 Hz),2.64-2.50 (m,1H),2.37- 2.23 (m,1H),1.38-1.37 (d,9H,J = 6 Hz),1.08-1.06 (m,3H); MS (ESI) m/z: 365.20 [M+H]+. HR-MS(ESI): m/z [M+H] calcd. for C19H28N2O5: 365.2071; found: 365.2066.
2.2. Synthesis of (R)-4-benzyl-7-methyl-1,4-diazepane-2,5-dione (4)
A solution of compound 3 (15.93 g,43.74 mmol) in 10 mL EtOAc was added 150 mL 45% HCl/EtOAc and the reaction was stirred for 4 h. The solvents were removed by rotary evaporation,and the residue was basified with saturated aqueous NaHCO3,and extracted with CH2Cl2. The organic extracts were concentrated. The residue was dissolved in 150 mL of dehydrated MeOH, treated with CH3ONa (2.84 g,52.49 mmol),and stirred at room temperature overnight (N2 protected,slightly exothermic). The reaction was cooled to room temperature and quenched with aqueous NH4Cl. Most of the solvent was removed and the reaction was then dumped into a separatory funnel containing 5% aqueous Na2CO3 and extracted with CH2Cl2 three times. The organic layers were combined,dried over MgSO4,and concentrated to provide compound 4 as a white solid 9.50 g in 94% yield. Analytical HPLC analysis carried out on Chiralpak AD column (4.6 mm × 250 mm) with 60% EtOH in hexanes (containing 0.1% diethylamine as a modifier),flow rate of 1 mL/min,indicated that intermediate (R)-4 was of >99% ee. Mp: 122-123 ℃. [α]D2533.5 (c 0.56,MeOH). 1H NMR (600 MHz,DMSO-d6): δ 7.77-7.76 (bd,1H,J = 6 Hz),7.33-7.25 (m,5H),4.59-4.53 (m,2H),4.10- 4.02 (m,2H),3.65-3.62 (m,1H),2.93-2.90 (m,1H),2.76-2.72 (m,1H), 1.14-1.13 (d,3H,J = 6 Hz); 13C NMR (150 MHz,DMSO-d6): δ 171.1, 168.4,138.1,128.9,128.0,127.7,53.1,50.6,46.5,40.5,23.3. MS (ESI) m/z: 233.10 [M+H]+. HR-MS(ESI): m/z [M+H] calcd. for C13H16N2O2: 233.1285; found: 233.1289.
2.3. Synthesis of (R)-1-benzyl-5-methyl-1,4-diazepane (6)
A solution of compound 4 (1.40 g,6.0 mmol) in 60 mL THF at 0 ℃ was treated with LiAlH4 (1.36 g,36.0 mmol) in batches. The reaction was slowly warmed to room temperature and stirred for another 4 h. The reaction was then cooled to -10 ℃ and was carefully quenched with 1.5 mL water,then NaOH (1.5 mL,15%) followed by an additional 4.5 mL of water. A portion of MgSO4was added and the mixture was stirred for 1 h before filtered. The filtrate was concentrated to provide light yellow oil 1.10 g in 88% yield. [α]D25 -5.9 (c 1.00,CHCl3),ee >99%,Analytical analysis was performed on Chrom Tech chiral-AGP column (150 mm × 4 mm) with 99% 1 mol/L ammonium dihydrogen phosphate and 1% acetonitrile,at flow rate of 0.5 mL/min with column temperature of 40 ℃. 1H NMR (600 MHz,DMSO-d6): δ 7.32-7.20 (m,5H),3.57 (s, 2H),3.48 (bs,1H),2.99-2.95 (m,1H),2.86-2.82 (m,1H),2.72-2.68 (m,1H),2.65-2.61 (m,1H),2.58-2.49 (m,3H),1.75-1.70 (m,1H), 1.46-1.41 (m,1H),1.01-1.00 (d,3H,J = 6 Hz); 13C NMR (150 MHz, DMSO-d6): δ 140.1,128.9,128.5,127.1,62.5,58.8,52.7,52.6,47.0, 37.5,23.9. MS (ESI) m/z: 205.10 [M+H]+. HR-MS(ESI): m/z [M+H] calcd. for C13H20N2: 205.1699; found: 205.1692.
2.4. Synthesis of (R)-(4-benzyl-7-methyl-1,4-diazepan-1-yl)(5-methyl-2-(2H-1,2,3-triazol-2-yl)phenyl)methanone (7)
To a solution of compound 6 (2.40 g,11.76 mmol),compound 5 (2.86 g,14.11 mmol),1-hydroxy-1H-benzotriazole (1.90 g, 14.11 mmol),and dry triethylamine (3.56 g,35.28 mmol) in 18 mL of dry DMF was added EDC hydrochloride (2.70 g, 14.11 mmol),and the reaction was stirred 2 h at room temperature. The reaction was partitioned between EtOAc and saturated aqueous NaHCO3,the layers were separated and the organic was added to aqueous citric acid stirring for 1 h. Water was added and the mixture was partitioned. Combined the water layers and added saturated aqueous Na2CO3 to regulate pH > 9,then extracted with three portions of EtOAc. The organic layers were combined,dried over MgSO4 and concentrated by rotary evaporation to provide compound 7 as a white power 4.30 g in 93% yield. Mp: 108-109 ℃, [α]D25-58.4 (c 1.01,MeOH). 1HNMR(600 MHz,DMSO-d6): δ 8.00- 7.76 (m,3H),7.37-7.17 (m,7H),4.40-4.09 (m,1H),3.63-3.48 (m, 2H),3.44-3.02 (m,3H),2.82-2.75 (m,1H),2.63-2.47 (m,1H), 2.63-2.14 (m,5H),2.02- 1.63 (m,2H),1.17-0.99 (m,3H); MS (ESI) m/z: 390.30 [M+H]+. HR-MS(ESI): m/z [M+H] calcd. for C23H27N5O: 390.2288; found: 390.2281.
2.5. Synthesis of (R)-(7-methyl-1,4-diazepan-1-yl)(5-methyl-2-(2H-1,2,3-triazol-2-yl)phenyl)methanone (9)
Compound 7 (5.86 g,15.05 mmol) was dissolved in 58 mL MeOH. After a portion of 10% Pd/C was added,the reaction was stirred for 4 h under H2 atmosphere at room temperature. The reaction was filtered through a pad of celite and the filtrate was concentrated to provide compound 9 as a white solid 4.01 g in 89% yield. Mp: 119-121 ℃,[a]D 26 -14.4 (c 1.00,MeOH)). 1H NMR (600 MHz,DMSO-d6): δ 8.24-8.02 (m,2H),7.88-7.29 (m,3H), 4.42-2.50 (m,7H),2.41 (s,3H),2.24-1.98 (m,2H),1.17-0.99 (m, 3H); 13C NMR (150 MHz,DMSO-d6): δ 168.6,138.3,136.9,134.1, 131.1,129.2,128.3,122.5,52.6,49.1,44.4,43.1,37.8,20.8,20.6. MS (ESI)m/z: 300.20 [M+H]+. HR-MS(ESI): m/z [M+H] calcd. for C16H21N5O: 300.1819; found: 300.1812.
2.6. Synthesis of suvorexant
To compound 8 (0.56 g,3 mmol) in 10 mL dry DMF was added TEA (0.91 g,9 mmol) and compound 9 (0.89 g,3 mmol),the mixture was stirred at 75 ℃ for 2 h. After cooling to room temperature,the reaction was diluted with EtOAc,washed with saturated aqueous NaHCO3,water,brine and dried over MgSO4. The residue was recrystallized from i-PrOH/EtOAc to provide a white solid 1.20 g in 90% yield. Mp: 149-150 ℃,[α]D25 -11.6 (c 1.00,MeOH). Analytical HPLC analysis carried out on a Chiralpak AD column (4.6 mm × 250 mm) with 60% EtOH in hexanes (containing 0.1% diethylamine as a modifier) at a flow rate of 1 mL/min,indicated that intermediate (R)-4 was of >99% ee. Mp: 153 ℃,[α]D25 -11.7 (c 1.00,MeOH) [ OPRD REF ],
Baxter, C. A.; Cleator, E.; Brands, K. M. J.; Edwards, J. S.; Reamer, R. A.; Sheen, F. J.; Stewart, G. W.; Strotman, N. A.; Wallace, D. J. (2011). “The First Large-Scale Synthesis of MK-4305: A Dual Orexin Receptor Antagonist for the Treatment of Sleep Disorder”.Organic Process Research & Development15 (2): 367–375. doi:10.1021/op1002853.
“Suvorexant: A Dual Orexin Receptor Antagonist for the Treatment of Sleep Onset and Sleep Maintenance Insomnia.”. Ann Pharmacother49: 477–483. Feb 9, 2015.doi:10.1177/1060028015570467. PMID25667197.
Label: BELSOMRA- Suvorexant Tablet, Film Coated”Label: BELSOMRA- Suvorexant Tablet, Film Coated.” DailyMed. Merck Sharp & Dohme Corp. & the U.S. National Library of Medicine, 01 Aug. 2014. Web. 29 Oct. 2014.
Jacobson, LH; Callander, GE; Hoyer, D (Nov 2014). “Suvorexant for the treatment of insomnia.”. Expert review of clinical pharmacology7 (6): 711–30.doi:10.1586/17512433.2014.966813. PMID25318834.
“Belsomra”. drugs.com. Retrieved 20 February 2015.
“U.S. Food and Drug Administration.” Drug Development and Drug Interactions: Table of Substrates, Inhibitors and Inducers. U.S. Food and Drug Administration, 27 Oct. 2014. Web. 30 Oct. 2014.
Suvorexant synthesis There are several ways, the following is a scaled-up process (OPRD, 2011, 15, 367). A compound with sulfur phosgene in ring closure to give 2,2 thiol group with oxalyl chloride to chlorine after conversion to give the intermediate 4 with a primary amine 3 attack, followed by Michael addition occurred with 5 6.6 mesylate de Boc protected After the reductive amination get 7, this is the racemic product. 7 8 after two crystallization with tartaric acid split to give 9 (> 97% ee).Triazole carboxylic acid 10 with 11 to give 12, 12 coupled after conversion to the acid chloride under basic conditions with pH 9 condensation Suvorexant.
Pantoprazole is a proton pump inhibitor drug used for short-term treatment of erosion and ulceration of the esophagus caused by gastroesophageal reflux disease.
Use
Pantoprazole is used for short-term treatment of erosion and ulceration of the oesophagus caused by gastroesophageal reflux disease. Initial treatment is generally of eight weeks’ duration, after which another eight week course of treatment may be considered if necessary. It can be used as a maintenance therapy for long term use after initial response is obtained.
Adverse effects
Antacid preparations such as pantoprazole work by suppressing the acid-mediated breakdown of proteins. This leads to an elevated risk of developing food and drug allergies due to undigested proteins passing into the gastrointestinal tract where sensitisation occurs. It is unclear whether this risk occurs with short-term or only long-term use.[1]
Nutrition: May reduce the absorption of important nutrients, vitamins and minerals, as well as medications, leaving users at increased risk for pneumonia.[2]
Cardiovascular: Increase in a chemical that suppresses the production of nitric oxide by 25% in humans, which have proven to relax and protect arteries and veins. Causes blood vessels to constrict, a development that could lead to a number of cardiovascular problems if continued for a prolonged period of time.[2]
Pharmacology
Wyeth pantoprazole 20mg.
Pantoprazole is metabolized in the liver by the cytochrome P450 system.[3] Metabolism mainly consists of demethylation by CYP2C19followed by sulfation. Another metabolic pathway is oxidation by CYP3A4. Pantoprazole metabolites are not thought to have any pharmacological significance. Pantoprazole is relatively free of drug interactions;[4] however, it may alter the absorption of other medications that depend on the amount of acid in the stomach, such as ketoconazole or digoxin. Generally inactive at acidic pH of stomach, thus it is usually given with a pro kinetic drug. Pantoprazole binds irreversibly to H+K+ATPase (proton pumps) and suppresses the secretion of acid. As it binds irreversibly to the pumps, new pumps have to be made before acid production can be resumed. The drug’s plasma half-life is about 2 hours.[5]
Pharmacokinetics
Absorption
Bioavailability: (oral, delayed release tablets), approximately 77%
Effect of food: (oral, delayed-release tablets), AUC and Cmax no effect, Tmax variable, absorption delayed, no net effect
Effect of food: (oral, for-delayed-release suspension), administer 30 minutes before a meal
Tmax, Oral, delayed-release suspension: 2 to 2.5 h
Tmax, Oral, delayed-release tablets: 2.5 h
Tmax, Oral, delayed-release tablets: 1.5 to 2 hours (pediatrics)
Distribution
Protein binding: about 98% to primarily albumin
Vd, extensive metabolizers (IV): approximately 11 L to 23.6 L
Vd, pediatrics (oral): 0.21 to 0.43 L/kg.
Metabolism
Hepatic; cytochrome P450 CYP2C19; minor metabolism from CYP3A4, 2D6, and 2C9
Excretion
Fecal: (oral or IV, normal metabolizers), 18%
Renal: (oral or IV, normal metabolizers), approximately 71%, none as unchanged
Dialyzable: no (hemodialysis)
Total body clearance: (IV) 7.6 to 14 L/hour.
Total body clearance: (oral, pediatrics) 0.18 to 2.08 L/h/kg
Elimination Half Life
Oral or IV, 1 hour
Oral or IV, slow metabolizers, 3.5 to 10 hours
Pediatrics, 0.7 to 5.34 hours
Availability
Pantoprazole was developed by Altana (owned by Nycomed) and was licensed in the USA to Wyeth (which was taken over by Pfizer). It was initially marketed under the brand name Protonix by Wyeth-Ayerst Laboratories and now is available as a generic. It is available by prescription in delayed-release tablets. It is also available for intravenous use.
On 24 December 2007, Teva Pharmaceutical released an AB-rated generic alternative to Protonix.[6] This was followed by generic equivalents from Sun Pharma and Kudco Pharma. Wyeth sued all three for patent infringement and launched its own generic version of Protonix with Nycomed.[7][8]
Pantoprazole is the international non-proprietary name of the chemical product 5-(difluoromethoxy)-2-[[(3,4-dimethoxy-2- pyridinyl)methyl]sulfmyl]-lH-benzimidazole of formula
Pantoprazole This product is an active ingredient used in the treatment of gastric ulcers, usually in the form of its sodium salt.
The product was described for the first time in European patent application EP-A-0166287 that also describes several processes for the preparation of products assignable to a general formula among which pantoprazole is to be found. The reaction sequences of these processes, applied precisely to the preparation of pantoprazole, are given in Scheme 1.
Scheme 1
In Scheme 1, the variables Y, Z, Z’ and Z” are leaving groups, for example atoms of halogen, and the variables M and M’ are atoms of alkali metals.
Austrian patent AT-B-394368 discloses another process based on a different route of synthetis, the reaction sequence of which is given in Scheme 2.
Pantoprazole Scheme 2
Nevertheless, this process has obvious drawbacks, since the methylation can take place not only in OH in the 4-position of the pyridine ring, but also in the nitrogen linked to a hydrogen of the benzimidazole ring, which can give place to mixtures of the desired product with the two possible methylated isomers of the benzimidazole compounds obtained, 3- methyl or 1 -methyl, which means that additional chromatographic purification steps are needed and the yields obtained are low.
PCT application WO97/29103 discloses another process for the preparation of pantoprazole, the reaction sequence of which is given in Scheme 3.
Scheme 3 As may be seen, different synthesis strategies have been proposed for the preparation of pantoprazole, some of them recently, which is an indication that the preparation of the product is still not considered to be sufficiently well developed, whereby there is still a need for developing alternative processes that allow pantoprazole to be prepared by means of simpler techniques and more accessible intermediate compounds and with good chemical yields.
EXAMPLES
Example 1. – Preparation of compound (IX)
47.5 ml (0.502 mol) of acetic anhydride were mixed with 1.65 g (0.0135 mol) of 4-dimethylaminopyridine, giving a transparent yellow solution which was heated to 65° – 70°C. This temperature was held by cooling since the reaction is exothermic. 25 g (0.1441 mol) of 2-methyl-3- methoxy-4-chloropyridine N-oxide (X) were added over a period of about 70 minutes. Once the addition was completed, the reaction was held at 65° – 70°C for a further 2h 20 minutes and after this time it was allowed to cool down to below 65°C and 90 ml of methanol were added gradually, while holding the temperature below 65°C. The resulting reaction mass was distilled at reduced pressure in a rotavap to remove the volatile components and the residue containing compound (IX) was used as such for the following reaction. Thin layer chromatography on silica gel 60 F254, eluting with CHCl3/MeOH (15: 1), showed a single spot at Rf – 0.82, indicating that the reaction has been completed.
Example 2. – Preparation of compound fVIII
(IX) (VIII)
11.5 ml methanol and 11.5 ml of water were added over the crude residue from Example 1 containing compound (IX), and thereafter, while holding the temperature to between 25° and 30°C with a water bath, the residual acetic acid contained in the crude residue was neutralized by the addition of 33% aqueous NaOH. Once the residual acid had been neutralized, 19 ml (0.2136 mol) of the 33% aqueous NaOH were added over 20 minutes, while holding the temperature to between 25° and 30°C, and, on completion of the addition, the hydrolysis reaction at pH 11.7 – 11.8 was held for 2h 30 minutes, to between 25° and 30°C. On completion of the reaction, the pH was adjusted to 7.0 – 7.5 by the addition of HC1 35%, while holding the temperature to 25°C. Thereafter, 50 ml of methylene chloride were added and, after stirring and allowing to rest, the phases were decanted. A further five extractions were carried out with 30 ml methylene chloride each and the pooled organic phases were dried with anhydrous sodium sulfate, were filtered and washed, and were evaporated at reduced pressure in a rotavap, providing a solid residue having a melting point around 73°C and containing compound (VIII). Thin layer chromatography on silica gel 60 F254, eluting with CHCl3/MeOH (15: 1), gave a main spot at Rf = 0.55, showing that the reaction was complete. The thus obtained crude residue was used as such in the following reaction.
Example 3. – Preparation of compound (VI)
24.5 g of the residue obtained in Example 2, containing approximately 0.142 mol of the compound 2-hydroxymethyl-3-methoxy-4-chloropyridine (VIII), were mixed with 0.5 ml of DMF and 300 ml of anhydrous methylene chloride, to give a brown solution which was cooled to 0° – 5°C in an ice water bath. Thereafter, a solution of 11.5 ml (0.1585 mol) of thionyl chloride in 50 ml of anhydrous methylene chloride was added over 20 minutes, while holding the above-mentioned temperature,. Once the addition was complete, the reaction was held at 0° – 5°C for a further 90 minutes and then 120 ml of water and NaOH 33% were added to pH 5 – 6, requiring approximately 29 ml of NaOH. The phases were then decanted and separated. The organic phase was extracted with a further 120 ml of water and the pooled aqueous phases were extracted with a further 4×25 ml of methylene chloride, in order to recover the greatest possible amount of product. The pooled organic phases were dried over anhydrous sodium sulfate, filtered and washed, and evaporated at reduced pressure in a rotavap, to give a residue containing the compound 2-chloromethyl-3- methoxy-4-chloropyridine (VI). Thin layer chromatography on silica gel 60 F254, eluting with CHCl3/MeOH (15:1), showed a main spot at Rf = 0.83, indicating that the reaction was complete. The thus obtained crude residue was used as such in the following reaction. Example 4. – Preparation of compound (III)
26.11 g of the residue obtained in the Example 3 containing approximately 0.136 mol of the compound 2-chloromethyl-3-methoxy-4- chloropyridine (VI) were mixed with 370 ml of methylene chloride, to give a brown solution over which were added, at 20° – 25°C, 29.3 g (0.136 mol) of 5-difluoromethoxy-2-mercaptobenzimidazole (VII) and 17.10 ml (0.136 mol) of tetramethylguanidine (TMGH). The mixture was stirred at this temperature for 2 hours, after which 450 ml of water were added, with the pH being held to between 9.5 and 10. Thereafter the phases were decanted and the organic phase was washed 5×50 ml of a IN NaOH aqueous solution and, thereafter, with 2×50 ml of water. The organic phase was treated with 50 ml of water and an amount of HC1 30% sufficient to adjust the pH to between 5 and 6. Thereafter, the phases were decanted, and the organic phase was dried over anhydrous sodium sulfate, was filtered and washed, and evaporated at reduced pressure in a rotavap, to give a solid residue of melting point 64° – 73 °C that contains the compound (III). Thin layer chromatography on silica gel 60 F254, eluting with CHCl3/MeOH (15: 1), presented a main spot at Rf = 0.52. Yield 82%. The thus obtained compound 5-(difluoromethoxy)-2-[[(3-methoxy-4-chlorine-2 pyridinyl)methyl]mercapto]- lH-benzimidazole (III) was used as such in the following reaction Example 5. – Preparation of compound (IV)
25.8 g (0.0694 mol) of the compound (III) obtained in the Example 4 were mixed with 88 ml of methanol, to give a brown solution to which 3.7 ml of water, 0.99 g of ammonium molybdate and 0.78 g of sodium carbonate were added. The system was cooled to 0°C – 5°C, 3.4 ml (0.0756 mol) of 60% hydrogen peroxide were added, and the reaction mixture was held at 0°C – 5°C for 1 – 2 days, the end point of the reaction being checked by thin layer chromatography on silica gel 60 F254, eluting with CHCl3/MeOH (15: l).
During the reaction the presence of hydrogen peroxide in the reaction medium was controlled by testing with potassium iodide, water and starch. When effected on a sample containing hydrogen peroxide, it provides a brown-black colour. If the assay is negative before the chromatographic control indicates completion of the reaction, more hydrogen peroxide is added.
On completion of the reaction, 260 ml of water were added, the system was cooled to 0°C – 5°C again and the mixture was stirred for 2 hours at this temperature. The solid precipitate was filtered, washed with abundant water, and dried at a temperature below 60°C, to give 5-(difluoromethoxy)-2-[[(3- methoxy-4-chlorine-2-pyridinyl)methyl]sulfinyl]-lH-benzimidazole (IV), melting point 130° – 136°C, with an 83.5% yield. Thin layer chromatography on silica gel 60 F254, eluting with CHCl3/MeOH (15: 1), gave a main spot at Rf = 0.5.
Compound (IV) can be purified, if desired, by the following crystallization method:
5 g of crude product was suspended in 16 ml of acetone and was heated to boiling until a dark brown solution was obtained. Thereafter the thus obtained solution was allowed to cool down to room temperature and then was then chilled again to -20°C, at which temperature the mixture was held for 23 hours without stirring. Thereafter the solid was filtered and washed with 6×4 ml of acetone chilled to -20°C. Once dry, the resulting white solid weighed 2.73 g, had a point of melting of 142°C and gave a single spot in thin layer chromatography. The IR spectrum of the compound on KBr is given in Figure 1.
The acetonic solution comprising the mother liquors of filtration and the washes was concentrated to a volume of 20 ml and a further 5 g of crude compound were added. The above described crystallization process was repeated to obtain a further 4.11 g of purified product of characteristics similar to the previous one.
The acetonic solution from the previous crystallization was concentrated to a volume of 17 ml and a further 4 g of crude compound were added. The above described crystallization process was repeated to obtain a further 2.91 g of purified product of similar characteristics to the previous ones.
The acetonic solution from the previous crystallization was concentrated to a volume of 15 ml and a further 4 g of crude compound were added. The above described crystallization process was repeated to obtain a further 3.3 g of purified product of similar characteristics to the previous ones.
The acetonic solution from the previous crystallization was concentrated to a volume of 16 ml and a further 4.36 g of crude compound were added. The above described crystallization process was repeated to obtain a further 3.62 g of purified product of similar characteristics to the previous ones.
Finally, the acetonic solution from the previous crystallization was concentrated to a volume of 10 – 12 ml and held at -20°C for two days without stirring. Thereafter, the solid was filtered and washed with 5×3 ml of acetone chilled to -20°C. Once dry, the solid weighed 1.26 g and had similar characteristics to the previous ones.
The total yield of all the crystallizations was 80%.
Example 6. – Preparation of pantoprazole
12.95 g (0.0334 mol) of compound (IV) purified by crystallization of Example 5 were mixed with 38 ml of N,N-dimethylacetamide and thereafter 7.03 g (0.1003 mol) of potassium methoxide were added, while holding the temperature to between 20°C and 30°C, whereby a dark brown mixture was obtained. The system was held at approximately 25°C for about 23 hours, after which, once the reaction was complete, the pH was adjusted to 7 with the addition of 3.82 ml of acetic acid. The N,N-dimethylacetamide was removed at reduced pressure at an internal temperature of not more than 75°C. 65 ml of water and 50 ml of methylene chloride were added over the thus obtained residue, followed by decantation of the phases. Once the phases were decanted, the aqueous phase was extracted a with further 3×25 ml of methylene chloride, the organic phases were pooled and the resulting solution dried over anhydrous sodium sulfate, was filtered and washed, and evaporated at reduced pressure in a rotavap, to give a crude residue over which 55 ml of water were added, to give a suspension (if the product does not solidify at this point the water is decanted and a further 55 ml of water are added to remove remains of N,N-dimethylacetamide that hinder the solidification of the product). The solid was filtered and, after drying, 11.61 g of crude pantoprazole of reddish brown colour were obtained (Yield 90%). The thus obtained crude product was decoloured by dissolving the crude product in 150 ml of methanol, whereby a dark brown solution was obtained. 7.5 g of active carbon were added, while maintaining stirring for 45 minutes at 25°C – 30°C, after which the carbon was filtered out and the filter was washed. The methanol was then removed in the rotavap at reduced pressure, a temperature below 40°C. 10.33 g of a solid residue were obtained and were mixed with 14.9 ml of methylethylketone, and the suspension was heated to 45°C for about 10 minutes, after which it was cooled, first to room temperature and then to -20°C. This temperature was held over night and thereafter the solid was filtered, washed with 6×5 ml of methylethylketone chilled to -20°C. Once dry, 7.75 g of a white solid, melting point 140°C – 141 °C, were obtained. Thin layer chromatography on silica gel F254, eluting with CHCl3/MeOH (15: 1), gave a single spot at Rf =
0.41 and a IR spectrum corresponding identically with that of pantoprazole.
The ketonic solution comprising the mother liquors of filtration and the washes, was concentrated to 9.7 ml, was heated to 40°C, was held at this temperature for about five minutes and was then cooled, first to room temperature and then to -20°C, this temperature being held for 4 hours. At the end of this time, the solid was filtered and was washed with 4×2 ml of methylethylketone chilled to -20°C. Once dry, 0.42 g of a white solid of similar characteristics to the previous one was obtained.
The ketone solution from the previous treatment was concentrated to 3.1 ml, was heated to 40°C, was held to this temperature for about five minutes and then was cooled, first to room temperature and then to -20°C, this temperature being held for 4 hours. At the end of this time, the solid was filtered and was washed with 5×3 ml of methylethylketone chilled to – 20°C. Once dry, 0.41 g of a white-beige solid of similar characteristics to the previous one was obtained. The total yield, including purifications, was 67%.
If a whiter solid is desired, one or several washes can be carried with isopropyl acetate as follows: 6.6 g of pantoprazole from the methylethylketone treatment were suspended in 50 ml of isopropyl acetate. The system (white suspension) was stirred for about 30 minutes at 25°C, was then cooled to 0°C – 5°C, was stirred for about 15 minutes at this temperature and the solid was then filtered, was washed with 3×15 ml of isopropyl acetate. Once dry, 6.26 g of a pure white solid were obtained.
Trade Names
Country
Trade name
Manufacturer
Germany
Pantozol
Nycomed
Rifun
– “-
France
Eupantol
Altana
Inipomp
Sanofi-Aventis
United Kingdom
Protium
ALTANA
Italy
Pantekta
Abbott
Pantopan
Pharmacia
Pantork
Altana
USA
Protonix
Wyeth
Ukraine
Kontrolok
Nycomed Oranienburg GmbH, Germany
Nolpaza
Krka
Pultset
Nobel Ilach Sanayi ve Ticaret AS, Turkey
Proksium
JSC “Lubnyfarm”, Ukraine
various generic drugs
Formulations
ampoule 40 mg;
Tablets 40 mg
UV – spectrum
Conditions : Concentration – 1 mg / 100 ml
Solvent designation schedule
Methanol
Water
0.1 M HCl
0.1M NaOH
The absorption maximum
289 nm
291nm
Observed
decay
295 nm
391
346
–
418
ε
16600
14700
–
17700
IR – spectrum
Wavelength (μm)
Wavenumber (cm -1 )
NMR Spectrum
will be added
Links
EP 134 400 (Byk Gulden Lomberg; appl. 1.5.1984; CH-prior. 3.5.1983).
US 4,555,518 (Byk Gulden Lomberg; 26.11.1985; appl. 1.5.1984; CH-prior. 3.5.1983).
US 4,758,579 (Byk Gulden Lomberg; 19.7.1988; appl. 28.4.1987; CH-prior. 16.6.1984).
UV and IR Spectra. H.-W. Dibbern, RM Muller, E. Wirbitzki, 2002 ECV
NIST / EPA / NIH Mass Spectral Library 2008
Handbook of Organic Compounds. NIR, IR, Raman, and UV-Vis Spectra Featuring Polymers and Surfactants, Jr., Jerry Workman.Academic Press, 2000.
Handbook of ultraviolet and visible absorption spectra of organic compounds, K. Hirayama. Plenum Press Data Division, 1967.
[Dr. John Cooke, chair of Methodist Hospital’s cardiovascular services] [Houston Chronicle Health Zone dated Thursday, July 11, 2013 chron.com/refluxmeds] (Journal: Circulation)
Jump up^ Meyer, U A (1996). “Metabolic interactions of the proton-pump inhibitors lansoprazole, omeprazole and pantoprazole with other drugs”. European journal of gastroenterology & hepatology8 (Suppl 1): S21–25. doi:10.1097/00042737-199610001-00005.
Steinijans, V. W.; Huber, R.; Hartmann, M.; Zech, K.; Bliesath, H.; Wurst, W.; Radtke, H. W. (1996). “Lack of pantoprazole drug interactions in man: An updated review”. International Journal of Clinical Pharmacology and Therapeutics34 (6): 243–262. PMID8793611.
Terconazole is an anti-fungal medication, primarily used to treat vaginal fungal infections.
The synthesis of racemic terconazole [J. Heeres et al., J. Med . Chem . , 26 , 611 11983)] is similar. differing in the introduction of a 1 H- 1 , 2,4-triazol-1-yl substituent in place of 1H-imidazol-1-yl and in the nature of the phenol used in the last step of the synthetic sequence, which phenol is 1-methylethyl-4-(4- hydroxyphenyl)piperazme instead of 1-acetyl-4-(4-nydroxyphenyl)piperazine.
Example 20: (2S,4R) -(-)-1-[4-[[2-(2,4-dichlorophenyl)-2-[(1H-1,2,4-triazol-1-yl]methyl-1,3-dioxolane-4-yl]methoxy]phenyl]-4-(1-methylethyl)piperazine, (2S,4R) – (-)-terconazole.
This compound is prepared following the process described for (+)-torconazole, starting from (2S,4S)-(-)-IV (Ar = 2,4-dichlorophenyl, Y = N, R = CH3) (224 mg, 0.55 mmol), 4-(4-hydroxyphenyl)-1-(1-methylethyl)-piperazine (121 mg, 0.55 mmol), NaH (22.4 mg, 0.56 mmol) in 8 ml of DMSO. (2S,4R) -(-(-terconazole ((2S,4R)-V, Ar
= 2,4-dichlorophenyl, Y = N, Z = CH(CH3)2) is obtained as a white solid, m.p. 76-78ºC, [α]D20= -12.0 (c = 0.4.
CHCl3).
Example 17 : (2R,4S)-(+)-1-[4-[[2-(2,4-dichlorophenyl)- 2-[(1H-1,2,4-triazol-1-yl]methyl-1,3-dioxolane-4-yl]methyl]phenyl]-4-(1-methylethyl)piperazine, (2R,4S)-(+)-terconazole.
To a suspension of NaH (60-65% dispersion in paraffin, 36 mg, 0.90 mmol) in anhydrous DMSO (8 ml), 4-(4-hydroxyphenyl) -1 – ( 1-methyle thyl ) p iper az ine ( 193 mg , 0 . 88 mmol ) is added and the mixture is stirred for 1 hour at room temperature. Then, (2R,4R)-(+)-IV (Ar = 2,4-dichlorophenyl, Y = N, R = CH3 ) is added (180 mg, 0.44 mmol) and the mixture is heated at 80°C for 4 hours. The reaction mixture is allowed to cool to room temperature, diluted with water (20 ml) and extraoteo with CH2Cl2 (3 × 25 ml). The combined organic phases are washed with 5N NaOH (3 × 25 ml) and water (3 × 25 ml dried with Na2SO4 and the solvent is evaporated of: under vacuum. The oily residue thus obtained is crystallized from diisopropyl ether to give (2R,4S)-(+)-terconazole ((2R,4S)-V, Ar = 2,4-cichlorophenyl, Y = N, Z = CH(CH3)2) (140 mg, 59 % yield) as a white solid, m.p. 72-74’C, [α]D20 = + 11,05 (c = 0.4, CHCl3).
The U.S. Food and Drug Administration today approved Cologuard, the first stool-based colorectal screening test that detects the presence of red blood cells and DNA mutations that may indicate the presence of certain kinds of abnormal growths that may be cancers such as colon cancer or precursors to cancer.
Colorectal cancer primarily affects people age 50 and older, and among cancers that affect both men and women, it is the third most common cancer and the second leading cause of cancer-related death in the United States, according to the Centers for Disease Control and Prevention (CDC). Colorectal cancer screening is effective at reducing illness and death related to colon cancer. The CDC estimates that if everyone age 50 or older had regular screening tests as recommended, at least 60 percent of colorectal cancer deaths could be avoided.
Colorectal cancer occurs in the colon (large intestine) or rectum (the passageway that connects the colon to the anus). Most colorectal cancers start as abnormal raised or flat tissue growths on the wall of the large intestine or rectum (polyps). Some very large polyps are called advanced adenomas and are more likely than smaller polyps to progress to cancer.
Using a stool sample, Cologuard detects hemoglobin, a protein molecule that is a component of blood. Cologuard also detects certain mutations associated with colorectal cancer in the DNA of cells shed by advanced adenomas as stool moves through the large intestine and rectum. Patients with positive test results are advised to undergo a diagnostic colonoscopy.
“This approval offers patients and physicians another option to screen for colorectal cancer,” said Alberto Gutierrez, Ph.D., director of the Office of In Vitro Diagnostics and Radiological Health at the FDA’s Center for Devices and Radiological Health. “Fecal blood testing is a well-established screening tool and the clinical data showed that the test detected more cancers than a commonly used fecal occult test.”
Today’s approval of the Cologuard does not change current practice guidelines for colorectal cancer screening. Stool DNA testing (also called “fecal DNA testing”) is not currently recommended as a method to screen for colorectal cancer by the United States Preventive Services Task Force (USPSTF). Among other guidelines, the USPSTF recommends adults age 50 to 75, at average risk for colon cancer, be screened using fecal occult blood testing, sigmoidoscopy, or colonoscopy.
The safety and effectiveness of Cologuard was established in a clinical trial that screened 10,023 subjects. The trial compared the performance of Cologuard to the fecal immunochemical test (FIT), a commonly used non-invasive screening test that detects blood in the stool. Cologuard accurately detected cancers and advanced adenomas more often than the FIT test. Cologuard detected 92 percent of colorectal cancers and 42 percent of advanced adenomas in the study population, while the FIT screening test detected 74 percent of cancers and 24 percent of advanced adenomas. Cologuard was less accurate than FIT at correctly identifying subjects negative for colorectal cancer or advanced adenomas. Cologuard correctly gave a negative screening result for 87 percent of the study subjects, while FIT provided accurate negative screening results for 95 percent of the study population.
Today the Centers for Medicare & Medicaid Services (CMS) issued a proposed national coverage determination for Cologuard. Cologuard is the first product reviewed through a joint FDA-CMS pilot program known as parallel review where the agencies concurrently review medical devices to help reduce the time between the FDA’s approval of a device and Medicare coverage. This voluntary pilot program is open to certain premarket approval applications for devices with new technologies and to medical devices that fall within the scope of a Part A or Part B Medicare benefit category and have not been subject to a national coverage determination.
“Parallel review allows the last part of the FDA process to run at the same time as the CMS process, cutting as many as six months from the time from study initiation to coverage,” said Nancy Stade, CDRH’s deputy director for policy. “The pilot program is ongoing, but we will apply what we have learned to improve the efficiency of the medical device approval pathway for devices that address an important public health need.”
“This is the first time in history that FDA has approved a technology and CMS has proposed national coverage on the same day,” said Patrick Conway, chief medical officer and deputy administrator for innovation and quality for CMS. “This parallel review represents unprecedented collaboration between the two agencies and industry and most importantly will provide timely access for Medicare beneficiaries to an innovative screening test to help in the early detection of colorectal cancer.”
CMS proposes to cover the Cologuard test once every three years for Medicare beneficiaries who meet all of the following criteria:
age 50 to 85 years,
asymptomatic (no signs or symptoms of colorectal disease including but not limited to lower gastrointestinal pain, blood in stool, positive guaiac fecal occult blood test or fecal immunochemical test), and
average risk of developing colorectal cancer (no personal history of adenomatous polyps, of colorectal cancer, or inflammatory bowel disease, including Crohn’s Disease and ulcerative colitis; no family history of colorectal cancers or an adenomatous polyp, familial adenomatous polyposis, or hereditary nonpolyposis colorectal cancer).
Cologuard is manufactured by Exact Sciences in Madison, Wisconsin.

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....



















































































































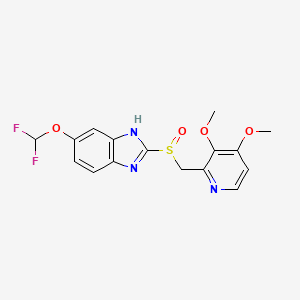

















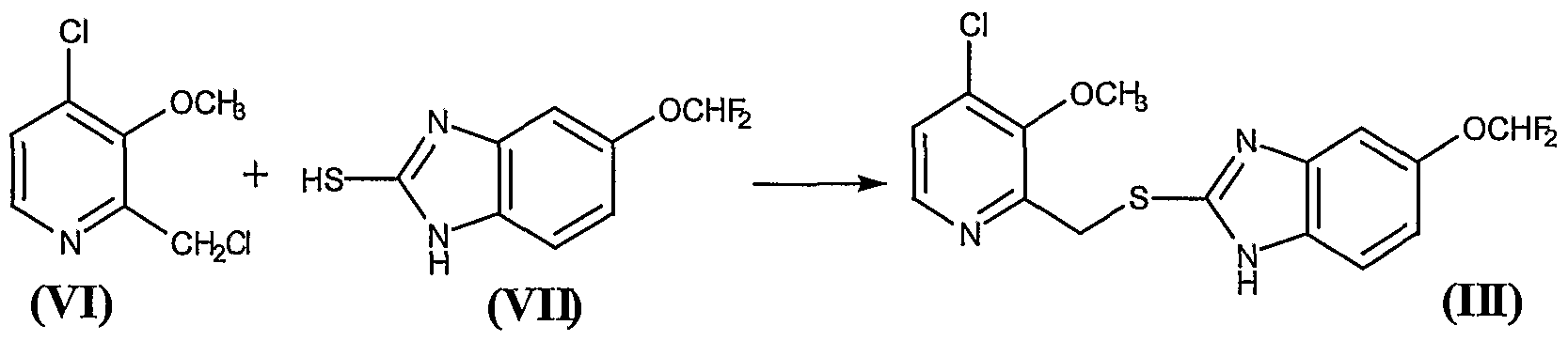





















{kind=link}