
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Tomensides A–D, new antiproliferative phenylpropanoid sucrose esters from Prunus tomentosa leaves…..might be valuable source for new potent anticancer drug candidates.

http://www.sciencedirect.com/science/article/pii/S0960894X14003540
Volume 24, Issue 11, 1 June 2014, Pages 2459–2462
Department of Natural Products Chemistry, Shenyang Pharmaceutical University, Shenyang 110016, PR China
To search for novel cytotoxic constituents against cancer cells as lead structures for drug development, four new 3-phenylpropanoid-triacetyl sucrose esters, named tomensides A–D (1–4), and three known analogs (5–7) were isolated from the leaves of Prunus tomentosa. Their structures were elucidated by spectroscopic analyses (1D, 2D NMR, CD and HRESIMS). The cytotoxic activities of all isolates against four human cancer cell lines (MCF-7, A549, HeLa and HT-29) were assayed, and the results showed that these isolates displayed stronger inhibitory activities compared with positive control 5-fluorouracil. Tomenside A (1) was the most active compound with IC50 values of 0.11–0.62 μM against the four tested cell lines. The structure–activity relationship (SAR) of the isolates was also discussed. The primary screening results indicated that these 3-phenylpropanoid-triacetyl sucrose esters might be valuable source for new potent anticancer drug candidates.
DY 268 as novel and potent antagonists of farnesoid X receptor

Farnesoid X receptor (FXR, NRIH4) plays a major role in the control of cholesterol metabolism. This suggests that antagonizing the transcriptional activity of FXR is a potential means to treat cholestasis and related metabolic disorders. Here we describe the synthesis, biological evaluation, and structure–activity relationship (SAR) studies of trisubstituted-pyrazol carboxamides as novel and potent FXR antagonists. One of these novel FXR antagonists, 4j has an IC50 of 7.5 nM in an FXR binding assay and 468.5 nM in a cell-based FXR antagonistic assay. Compound 4j has no detectable FXR agonistic activity or cytotoxicity. Notably, 4j is the most potent FXR antagonist identified to date; it has a promising in vitro profile and could serve as an excellent chemical tool to elucidate the biological function of FXR.
Bioorganic & Medicinal Chemistry
Identification of trisubstituted-pyrazol carboxamide analogs as novel and potent antagonists of farnesoid X receptor
Original Research Article
Pages 2919-2938
Donna D. Yu, Wenwei Lin, Barry M. Forman, Taosheng Chen
Volume 22, Issue 11, Pages 2907-3066 (1 June 2014)
Lupin forms joint venture with Yoshindo
Lupin forms joint venture with Yoshindo
Indian pharmaceutical company Lupin is to create a new biosimilars company in a joint venture with Japanese pharmaceuticals company Yoshindo. The new company, to be called YL Biologics (YLB), will be jointly managed by both partners and will develop biosimilars including regulatory filings and…
read at
![]()
Fast-tracking new treatment for childhood cancer
An untreated neuroblastoma cell.
Children fighting a life-threatening form of cancer could be treated with a revolutionary anti-cancer therapy as early as next year, following the formation of a research alliance to fast-track development of a medicine pioneered by Australian researchers.
The Children’s Oncology Drug Alliance (CODA) unites the research and resources of UNSW Australia and its commercialisation arm, NewSouth Innovations, childhood cancer research charity The Kids’ Cancer Project, ASX-listed Australian biotechnology-company Novogen, and Nationwide Children’s Hospital, Columbus, Ohio, to accelerate development of a treatment purpose-built for neuroblastoma – the most common form of cancer in infancy.
Currently there is no medicine approved to treat neuroblastoma, a cancer that affects up to 100 children in Australia and around 650 in the United States each year. Childhood cancers – which claim the lives of three Australian children every week – are currently treated with chemotherapies that have been developed for adults…
View original post 668 more words
AZD 9291, Osimertinib, Third-generation, oral, irreversible, selective epidermal growth factor receptor (EGFR) inhibitor for Non-small cell lung cancer (NSCLC)

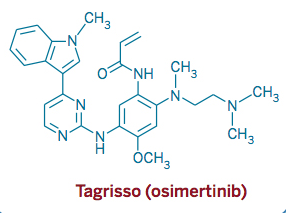
AZD 9291, Osimertinib
2-Propenamide, N-[2-[[2-(dimethylamino)ethyl]methylamino]-4-methoxy-5-[[4-(1-methyl-1H-indol-3-yl)-2-pyrimidinyl]amino]phenyl]-
N-(2-((2-(dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((4-(1-methyl-1H-indol-3-yl)pyrimidin-2-yl)amino)phenyl)acrylamide.
cas :1421373-65-0, 1421373-66-1(mesylate salt)
UPDATE…………FDA APPROVED NOV2015
EU …… 3 FEB 2016 APPROVED
03 February 2016
AstraZeneca today announced that the European Commission (EC) has granted conditional marketing authorisation for TAGRISSO™ (AZD9291, osimertinib) 80mg once-daily tablets for the treatment of adult patients with locally advanced or metastatic epidermal growth factor receptor (EGFR) T790M mutation-positive non-small cell lung cancer (NSCLC).
Osimertinib is indicated for patients with T790M mutation-positive NSCLC, irrespective of previous treatment with a
ASTRAZENECA
Astrazeneca Ab, Astrazeneca Uk Limited

Mechanism of Action: Third-generation, oral, irreversible, selective epidermal growth factor receptor (EGFR) inhibitor
Non-small cell lung cancer (NSCLC)
AZD-9291 M. Wt: 499.61
AZD-9291 Formula: C28H33N7O2
AZD9291, a third-generation orally irreversible epidermal growth factor receptor (EGFR) inhibitor, is under development by British drug maker AstraZeneca for the treatment of patients with metastatic EGFR T790M mutation-positive non-small cell lung cancer (NSCLC).
Lung cancer is the major cause of cancer death in the world while non small cell lung cancer (NSCLC) accounts approx. 85% of all lung cancer diagnosis. Approximately 50% of non–small cell lung cancer (NSCLC) patients who develop resistance to inhibitors of the epidermal growth factor receptor (EGFR) have acquired a second mutation, T790M. There are currently no approved treatments for patients who develop a T790 mutation.

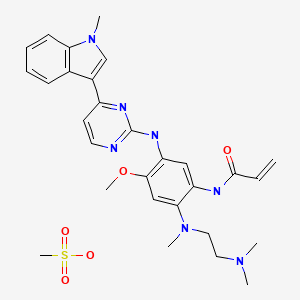
AZD9291 mesylate

Osimertinib (previously known as mereletinib[2] and AZD9291; trade name Tagrisso) is a third-generation epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI) drug[3][4] developed by AstraZeneca Pharmaceuticals – for mutated EGFR cancers.

Approvals and indications
In November 2015, after a Priority Review, the US FDA granted accelerated approval to osimertinib for the treatment of metastaticepidermal growth factor receptor (EGFR) T790M mutation-positive non-small cell lung cancer (NSCLC), as detected by an FDA-approved test, which has progressed on or after EGFR tyrosine kinase inhibitor (TKI) therapy.[5][6]

The FDA approval made reference to two clinical trials, in which an EGFR T790M mutation was confirmed by a Cobas EGFR mutation test; osimertinib was given as 80 mg once daily.[1][7]
AZD-9291 is a third-generation EGFR inhibitor, showed promise in preclinical studies and provides hope for patients with advanced lung cancers that have become resistant to existing EGFR inhibitors. AZD9291 is highly active in preclinical models and is well tolerated in animal models. It inhibits both activating and resistant EGFR mutations while sparing the normal form of EGFR that is present in normal skin and gut cells, thereby reducing the side effects encountered with currently available medicines

Synthesis of AZD9291,

CLICK ON IMAGES FOR CLEAR VIEW
WO 2013014448
http://www.google.com/patents/WO2013014448A1?cl=en
Example 27: V-f5-{[5-Cvano-4-flH-indol-3-yl)pyrimidin-2-yllamino}-4-methoxy-2-{4- methylpiperazin-l-yl}phenyl)prop-2-enamide
Acryloyl chloride (0.100 mL, 1M in THF, 0.1 mmol) was added dropwise to a fine slurry of 2- {[5-amino-2-methoxy-4-(4-methylpiperazin- 1 -yl)phenyl] amino} -4-(lH-indol-3- yl)pyrimidine-5-carbonitrile (Intermediate 99, 47 mg, 0.10 mmol) and DIPEA (0.027 mL, 0.16 mmol) in THF (2 mL) at -10°C over a period of 2 minutes under N2.The mixture was then stirred at 0°C for 10 minutes then allowed to warm to r.t. over 20 minutes. The mixture was then cooled again to -10°C and further acryloyl chloride (0.06 mL, 1M in THF, 0.06 mmol) was added dropwise. The mixture was stirred at 0°C for a further 10 minutes, then allowed to warm to r.t. over 20 minutes. The mixture was then concentrated in vacuo and the resulting reside was dissolved in CH2C12 (2 mL). This solution was washed with sat. NaHC03 (1 mL), dried (MgS04) and concentrated in vacuo. Purification by FCC, eluting with 1.5-7% 7N methanolic ammonia in CH2C12 gave a residue that was washed with CH3OH (0.1 mL) and dried in air to give the title compound (1 lmg, 20%) as a cream crystalline solid; 1H NMR: 2.28 (3H, s), 2.54-2.65 (4H, m), 2.93 (4H, s), 3.75 (3H, s), 5.71 (1H, d), 6.18 (1H, d), 6.64 (1H, dd), 6.91 (2H, m), 7.18 (1H, s), 7.47 (1H, d), 8.02 (1H, s), 8.52 (1H, s), 8.67 (1H, s), 9.04 (1H, s), 9.40 (1H, s), 11.99 (1H, s); m/z: ES+ MH+ 509. Example 28: V-f2-{2-Dimethylaminoethyl-methylamino}-4-methoxy-5-{[4-fl- methylindol-3-yl)pyrimidin-2-yllamino}phenyl)prop-2-enamide

A solution of acryloyl chloride (34.5 mg, 0.38 mmol) in CH2C12 (1 mL) was added dropwise to a stirred mixture of N;-(2-dimethylaminoethyl)-5-methoxy-N;-methyl-N¥-[4- (l-methylindol-3-yl)pyrimidin-2-yl]benzene-l,2,4-triamine (Intermediate 100, 170 mg, 0.38 mmol) and DIPEA (0.073 mL, 0.42 mmol) in CH2C12 (5 mL), which was cooled in an ice/water bath. The mixture was stirred for 1.5h and then diluted with CH2C12 (25 mL) and washed with sat.NaHCOs (50 mL). The aqueous washes were extracted with CH2C12 (2 x 25 mL). The combined organic solutions were dried (MgSC^) and concentrated in vacuo. Purification by FCC, eluting with 0-4% 7N methanolic ammonia in CH2C12 gave the title compound (75 mg, 39%) as a cream solid after trituration with diethyl ether; 1H NMR: 2.21 (6H, s), 2.29 (2H, t), 2.72 (3H, s), 2.89 (2H, t), 3.86 (3H, s), 3.92 (3H, s), 5.77 (1H, dd), 6.27 (1H, dd), 6.43 (1H, dd), 7.04 (1H, s), 7.15 (1H, t), 7.20-7.27 (2H, m), 7.53 (1H, d), 7.91 (1H, s), 8.24 (1H, d), 8.33 (1H, d), 8.68 (1H, s), 9.14 (1H, s), 10.22 (1H, s); m/z: ES+ MH+ 500.42.
Example 28 (Alternative synthesis 1): V-f2-{2-Dimethylaminoethyl-methylamino}-4- methoxy-5-{[4-(l-methylindol-3-yl)pyrimidin-2-yllamino}phenyl)prop-2-enamide

To a stirred solution of 3-chloro-N-[2-[2-dimethylaminoethyl(methyl)amino]-4-methoxy- 5-[[4-(l-methylindol-3-yl)pyrimidin-2-yl]amino]phenyl]propanamide (Intermediate 174, 31.5 g, 58.76 mmol) in acetonitrile (310 mL) was added triethylamine (17.84 g, 176.28 mmol) at r.t. The resulting mixture was heated to 80°C for 6h then cooled to r.t.. Water (130 mL) was then added and the mixture stirred for 12h. The mixture was then filtered, washed with a mixture of water and acetonitrile (160 mL, 1 : 1) and dried at 50°C for overnight to give the title compound (19.2 g, 94%) as a solid form identified herein as polymorphic form D. 1H NMR: 2.69 (3H, s), 2.83 (6H, d), 3.35 (4H, s), 3.84 (3H, s), 3.91 (3H, s), 5.75 (IH, d), 6.28 (IH, d), 6.67 (IH, dd), 7.05-7.23 (2H, m), 7.29 (IH, t), 7.43 (IH, d), 7.56 (IH, d), 8.21 (2H, s), 8.81 (IH, s), 9.47 (IH, s), 9.52 (IH, s), m/z: ES+ MH+ 500.26.
Example 28 (Alternative synthesis 2): V-f2-{2-Dimethylaminoethyl-methylamino}-4- methoxy-5-{[4-(l-methylindol-3-yl)pyrimidin-2-yllamino}phenyl)prop-2-enamide

To a stirred solution of N1-(2-dimethylaminoethyl)-5-methoxy-N1-methyl-N4-[4-(l- methylindol-3-yl)pyrimidin-2-yl]benzene-l,2,4-triamine (Intermediate 100, 10 g, 21.32 mmol) in THF (95 mL) and water (9.5 mL) at 0°C was added the 3-chloropropanoyl chloride (3.28 g, 25.59 mmol). The mixture was stirred at r.t. for 15 minutes then NaOH (3.48 g, 85.28 mmol) was added. The resulting mixture was heated to 65°C for lOh. The mixture was then cooled to r.t. and CH3OH (40 mL) and water (70 mL) were added. The resulting mixture was stirred overnight. The resulting solid was collected by filtration, washed with water (25 mL) and dried at 50°C for 12h to give the title compound (7.0 g, 94%) as a solid form identified herein as polymorphic Form D.1H NMR: 2.69 (3H, s) 2.83 (6H, d) 3.35 (4H, s) 3.84 (3H, s) 3.91 (3H, s) 5.75 (IH, d) 6.28 (IH, d) 6.67 (IH, dd) 7.05-7.23 (2H, m) 7.29 (IH, t) 7.43 (IH, d) 7.56 (IH, d) 8.21 (2H, s) 8.81 (IH, s) 9.47 (IH, s) 9.52 (IH, s) ES+ MH+ 500.26.
Example 28A: V-f2-{2-Dimethylaminoethyl-methylamino}-4-methoxy-5-{[4-fl- methylindol-3-yl)pyrimidin-2-yll amino}phenyl)prop-2-enamide mesylate salt

Procedure 1: To a stirred solution of N-[2-[2-dimethylaminoethyl(methyl)amino]-4- methoxy-5 – [[4-( 1 -methylindol-3 -yl)pyrimidin-2-yl] amino]phenyl]prop-2-enamide
(Example 28, 20 g, 36.63 mmol) in ethanol (120 mL) and EtOAc (80 mL) at 70°C was added methane sulfonic acid (3.59 g, 36.63 mmol) as a solution in EtOAc (40 mL). The resulting mixture was stirred for 1.5h. The resulting solid was collected by filtration and dried at 80°C under vacuum overnight to give the title salt (20.5 g, 94%) in a solid form defined herein as polymorphic Form B for this salt.
Procedure 2: To a stirred solution of N-[2-[2-dimethylaminoethyl(methyl)amino]-4- methoxy-5 – [[4-( 1 -methylindol-3 -yl)pyrimidin-2-yl] amino]phenyl]prop-2-enamide
(Example 28, 5 g, 9.11 mmol) in acetone (45.5 mL) and water (4.55 mL) at 50°C was added methane sulfonic acid (0.893 g, 9.11 mmol) as a solution in acetone (4.55 mL). The resulting mixture was stirred for 1.5h. The resulting solid was collected by filtration and dried at 80°C under vacuum overnight to give the title salt (4.9 g, 94%) in a solid form defined herein as polymorphic Form B for this salt; ΧΗ NMR (acetone-ii6): 2.72 (3H, s), 2.96 (3H, s), 3.01 (6H, s), 3.58 (3H, t), 3.87-3.90 (7H, m), 5.76 (1H, dd), 6.38-6.53 (2H, m), 7.12 (1H, t), 7.20 (1H, t), 7.29 (1H, s), 7.40 (2H, t), 8.07-8.16 (3H, m), 8.56 (1H, s), 9.30 (1H, s), 9.60 (1H, s), 9.66 (1H, s ); m/z: ES+ MH+ 500.26.
Procedure 3: Polymorphic Form A of N-(2-{2-dimethylaminoethyl-methylamino}-4- methoxy-5 – { [4-( 1 -methylindol-3 -yl)pyrimidin-2-yl] amino } phenyl)prop-2-enamide mesylate salt was prepared in a similar manner as described above on a ~50 mg scale, except that acetonitrile was used as the solvent. Specifically, ~9.6mg methanesulfonic acid was dissolved into a minimum volumn of acetonitrile. ~50 mg N-(2- {2-dimethylamino- ethyl-methylamino } -4-methoxy-5 – { [4-( 1 -methylindol-3 -yl)pyrimidin-2-yl] amino } phenyl)- prop-2-enamide was also dissolved into a minimum volume of acetonitrile and then the resulting solution was added to the methanesulfonic acid solution. Formation of a solid resulted upon addition. This solid was collected by filtration and was air-dried and then analysed. The particular solid form produced in this experiment was designated as
Polymorphic Form A for this salt.
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
N-(2-{2-dimethylaminoethyl-methylamino}-4-methoxy-5-{[4-(1-methylindol-3-yl)pyrimidin-2-yl]amino}phenyl)prop-2-enamide
|
|
| Clinical data | |
| Trade names | Tagrisso |
| Routes of administration |
Oral tablets |
| Pharmacokinetic data | |
| Protein binding | probably high[1] |
| Metabolism | oxidation (CYP3A) |
| Biological half-life | 48 hours |
| Excretion | feces (68%), urine (14%) |
| Identifiers | |
| CAS Number | 1421373-65-0 |
| PubChem | CID 71496458 |
| ChemSpider | 31042598 |
| UNII | 3C06JJ0Z2O |
| ChEBI | CHEBI:90943 |
| Chemical data | |
| Formula | C28H33N7O2 |
| Molar mass | 499.6 g/mol |
References
- “Tagrisso (osimertinib) Tablet, for Oral Use. Full Prescribing Information” (PDF). AstraZeneca Pharmaceuticals LP, Wilmington, DE 19850. Retrieved 16 November 2015.
- “Proposed INN: List 113” (PDF). International Nonproprietary Names for Pharmaceutical Substances (INN) 29 (2): 285. 2015. Retrieved16 November 2015.
- Ayeni D, Politi K, Goldberg SB (2015). “Emerging Agents and New Mutations in EGFR-Mutant Lung Cancer”. Clin. Cancer Res. 21 (17): 3818–20. doi:10.1158/1078-0432.CCR-15-1211. PMID 26169963.
- Tan CS, Gilligan D, Pacey S (2015). “Treatment approaches for EGFR-inhibitor-resistant patients with non-small-cell lung cancer”. Lancet Oncol. 16 (9): e447–59. doi:10.1016/S1470-2045(15)00246-6. PMID 26370354.
- U.S. Food and Drug Administration. Hematology/Oncology (Cancer) Approvals & Safety Notifications. [1] Xu M, Xie Y, Ni S, Liu H (2015). “The latest therapeutic strategies after resistance to first generation epidermal growth factor receptor tyrosine kinase inhibitors (EGFR TKIs) in patients with non-small cell lung cancer (NSCLC)”. Ann Transl Med 3 (7): 96.doi:10.3978/j.issn.2305-5839.2015.03.60. PMC 4430733. PMID 26015938.
- U.S. Food and Drug Administration. “Osimertinib”. [2]

AstraZeneca R&D Facility, Alderley Park, Cheshire, United Kingdom
Cheshire Map Showing Location of Alderley Park
District: Macclesfield
Easting: 384439 Northing: 374467
Latitude: 53.27 Longitude: -2.23
 = Alderley Park
= Alderley Park
| INN or syn | URL | CID | InChIKey |
| AZ11657312 | |||
| AZ876 | AZ12260493 | CID 11655079 | IVANYIPLGFVBGR-UHFFFAOYSA-N |
| AZ12419304 | |||
| AZ12609721 | |||
| AZ12823138 | |||
| AZ12971554 | CID 44537923 | GMKHQRCPNMGCIX-ZWKOTPCHSA-N | |
| AZ13483342 | |||
| AZD0328 | CID 9794392 | OCKIPDMKGPYYJS-ZDUSSCGKSA-N | |
| Saracatinib | AZD0530 | CID 10302451 | OUKYUETWWIPKQR-UHFFFAOYSA-N |
| Barasertib | AZD1152 | CID 11497983 | GBJVVSCPOBPEIT-UHFFFAOYSA-N |
| AZD1208 | CID 58423153 | MCUJKPPARUPFJM-UWCCDQBKSA-N | |
| AZD1236 | |||
| AZD1332 | CID 49831044 | LBVKEEFIPBQIMD-UHFFFAOYSA-N | |
| AZD1480 | CID 16659841 | PDOQBOJDRPLBQU-QMMMGPOBSA-N | |
| AZD1656 | CID 16039797 | FJEJHJINOKKDCW-INIZCTEOSA-N | |
| MK175 | AZD1775 | CID 24856436 | BKWJAKQVGHWELA-UHFFFAOYSA-N |
| AZD1981 | CID 11292191 | JWYIGNODXSRKGP-UHFFFAOYSA-N | |
| AZD2014 | CID 25262792 | JUSFANSTBFGBAF-IRXDYDNUSA-N | |
| Olaparib | AZD2281 | CID 23725625 | FDLYAMZZIXQODN-UHFFFAOYSA-N |
| AZD2624 | CID 23649245 | QYTBBBAHNIWFOD-NRFANRHFSA-N | |
| AZD2927 | CID 57345449 | GAHPWXLXWUVMIV-MRXNPFEDSA-N | |
| Lesogaberan | AZD3355 | CID 9833984 | WVTGPBOMAQLPCP-GSVOUGTGSA-O |
| AZD3463 | CID 56599293 | GCYIGMXOIWJGBU-UHFFFAOYSA-N | |
| AZD3857 | |||
| AZD4017 | CID 24946280 | NCDZABJPWMBMIQ-INIZCTEOSA-N | |
| AZD4320 | |||
| AZD4547 | CID 51039095 | NCDZABJPWMBMIQ-INIZCTEOSA-N | |
| AZD4877 | CID 10368812 | SMFXSYMLJDHGIE-UHFFFAOYSA-N | |
| AZD5363 | CID 25227436 | JDUBGYFRJFOXQC-KRWDZBQOSA-N | |
| AZD5582 | CID 49847690 | WLMCRYCCYXHPQF-ZVMUOSSASA-N | |
| TX4 | AZD5904 | CID 10264211 | RSPDBEVKURKEII-ZCFIWIBFSA-N |
| Selumetinib | AZD6244 | CID 10127622 | IAYGCINLNONXHY-LBPRGKRZSA-N |
| AZD6495 | |||
| AZD6738 | |||
| Lanicemine | AZD6765 | CID 3038485 | FWUQWDCOOWEXRY-UHFFFAOYSA-N |
| AZD7325 | CID 23581869 | KYDURMHFWXCKMW-UHFFFAOYSA-N | |
| AZD7762 | CID 11152667 | IAYGCINLNONXHY-LBPRGKRZSA-N | |
| AZD8055 | CID 25262965 | KVLFRAWTRWDEDF-IRXDYDNUSA-N | |
| AZD8186 | CID 52913929 | LMJFJIDLEAWOQJ-UHFFFAOYSA-N | |
| AZD8329 | CID 25006684 | XWBXJBSVYVJAMZ-UHFFFAOYSA-N | |
| AZD8529 | CID 25125217 | IPCYZQQFECEHLI-UHFFFAOYSA-N | |
| AZD8542 | CID 53344810 | SMQVBAGSZVHCJP-UHFFFAOYSA-N | |
| AZD8931 | CID 11488320 | DFJSJLGUIXFDJP-UHFFFAOYSA-N | |
| STAT3Rx | AZD9150 | ||
| AZD9291 | CID 71496458 | DUYJMQONPNNFPI-UHFFFAOYSA-N | |
| Alvelestat | AZD9668 | CID 46861623 | QNQZWEGMKJBHEM-UHFFFAOYSA-N |
| Zibotentan | ZD4054 | CID 9910224 | FJHHZXWJVIEFGJ-UHFFFAOYSA-N |
| Vandetanib | ZD6474 | CID 3081361 | UHTHHESEBZOYNR-UHFFFAOYSA-N |
INNs or synonyms are in the first column, the 2nd column is the AZ OI links, 3rd column the PubChem CID, followed by the InChIKey as the 4th column.
UPDATE
13 November 2015
One of fastest development programmes – from start of clinical trials to approval in just over two and a half years to meet unmet patient need
With objective response rate of 59% and duration of response of 12.4 months, TAGRISSO provides important new option for patients
AstraZeneca today announced that the US Food and Drug Administration (FDA) has approved TAGRISSO™ (AZD9291) 80mg once-daily tablets for the treatment of patients with metastatic epidermal growth factor receptor (EGFR) T790M mutation-positive non-small cell lung cancer (NSCLC), as detected by an FDA-approved test, who have progressed on or after EGFR tyrosine kinase inhibitor (TKI) therapy.
AZD9291 is the only approved medicine indicated for patients with metastatic EGFR T790M mutation-positive non-small cell lung cancer. This indication is approved under the FDA’s accelerated approval process based on tumour response rate and duration of response (DoR).
AZD9291 is an EGFR-TKI, a targeted cancer therapy, designed to inhibit both the activating, sensitising mutations (EGFRm), and T790M, a genetic mutation responsible for EGFR-TKI treatment resistance. Nearly two-thirds of NSCLC patients who are EGFR mutation-positive and experience disease progression after being treated with an EGFR-TKI develop the T790M resistance mutation, for which there have been limited treatment options.
Pasi A Jänne MD, PhD, Director, Lowe Center for Thoracic Oncology at Dana-Farber Cancer Institute, Scientific Director, Belfer Center for Applied Cancer Science and Professor of Medicine, Harvard Medical School, said: “In the AURA clinical studies, AZD9291 has demonstrated compelling early efficacy and tolerability in patients with EGFRm T790M metastatic non-small cell lung cancer. This treatment has the potential to become the standard of care for patients living with EGFRm T790M non-small cell lung cancer. The accelerated approval of AZD9291 highlights its clinical promise for a targeted group of patients and gives healthcare providers an important new option.”
Pascal Soriot, Chief Executive Officer, AstraZeneca, said: “The FDA approval of TAGRISSO marks an important milestone for lung cancer patients who urgently need new treatment options. We have built on our heritage in this area and acted on the breakthrough clinical evidence to ensure this next-generation medicine reaches patients in record time. As we advance our comprehensive lung cancer portfolio, we have the opportunity to treat greater numbers of patients across all stages of this disease through precision medicines, immunotherapies and novel combinations.”
AstraZeneca has collaborated with Roche to develop the cobas® EGFR Mutation Test v2 as the companion diagnostic for AZD9291. The cobas® EGFR Mutation Test v2 is intended to identify a range of EGFR mutations in patients with non-small cell lung cancer, including T790M.
AZD9291 was granted Fast Track, Breakthrough Therapy, Priority Review and Accelerated Approval status by the FDA. In Europe and Japan, AZD9291 was granted Accelerated Assessment and Priority Review status respectively. Interactions with regulatory authorities in the rest of the world are ongoing.
The FDA approval of AZD9291 is based on data from the two AURA Phase II studies (AURA extension and AURA2) which demonstrated efficacy in 411 EGFRm T790M NSCLC patients that had progressed on or after an EGFR TKI. In those trials, overall objective response rate ((ORR) a measurement of tumor shrinkage) was 59% (95% CI: 54% to 64%). In a supportive Phase I study in 63 patients, ORR was 51% and median duration of response was 12.4 months.
The AZD9291 tolerability profile showed that no individual severe grade 3+ adverse events occurred at ≥ 3.5%.The most common adverse events were generally mild to moderate and included diarrhoea (42% all grades; 1.0% Grade 3/4), rash (41% all grades; 0.5% Grade 3/4), dry skin (31% all grades; 0% Grade 3/4), and nail toxicity (25% all grades; 0% Grade 3/4). There are no contraindications for AZD9291. Warnings and precautions include interstitial lung disease, QT interval prolongation, cardiomyopathy and embryofoetal toxicity.
AZD9291 Development Programme
AZD9291 is being studied in the confirmatory trial, AURA3, an open label, randomised Phase III study designed to assess the efficacy and safety of AZD9291 versus platinum-based doublet chemotherapy in patients with EGFR T790M positive, locally advanced, or metastatic NSCLC who have progressed following prior therapy with an EGFR-TKI. AZD9291 is also being investigated in the adjuvant setting and in the metastatic first-line setting, including in patients with brain metastases, as well as in combination with other compounds.
NOTES TO EDITORS
About Non-Small Cell Lung Cancer
Lung cancer is the leading cause of cancer death among both men and women, accounting for about one-third of all cancer deaths, more than breast, prostate and colorectal cancers combined. Lung cancer has a five-year survival rate that is less than 20%. Approximately 85% of all lung cancers in the US are NSCLC; 10% to 15% of these are EGFR mutation-positive. Approximately two-thirds of patients treated with EGFR TKI therapy will acquire resistance related to the T790M mutation.
About AZD9291
AZD9291 80mg once-daily tablet is the first medicine indicated for the treatment of patients with metastatic epidermal growth factor receptor (EGFR) T790M mutation-positive non-small-cell lung cancer (NSCLC), as detected by an FDA-approved test, who have progressed on or after EGFR tyrosine kinase inhibitor (TKI) therapy. Non-clinical in vitro studies have demonstrated that AZD9291 has high potency and inhibitory activity against mutant EGFR phosphorylation across the range of clinically relevant EGFRm and T790M mutant NSCLC cell lines with significantly less activity against EGFR in wild-type cell lines.
Osimertinib has recently been published by the World Health Organisation (WHO) as the proposed International Non-proprietary Name (INN) for AZD9291, and may become formally adopted during November 2015. In the US, the American Medical Association accepted osimertinib as the United States Adopted Name (USAN).
About AstraZeneca in Oncology
Oncology is a therapeutic area in which AstraZeneca has deep-rooted heritage. It will be potentially transformational for the company’s future, becoming the sixth growth platform. Our vision is to help patients by redefining the cancer treatment paradigm and one day eliminate cancer as cause of death. By 2020, we are aiming to bring six new cancer medicines to patients.
Our broad pipeline of next-generation medicines is focused on four main disease areas – lung, ovarian, breast, and hematological cancers. These are being targeted through four key platforms – immuno-oncology, the genetic drivers of cancer and resistance, DNA damage repair and antibody drug conjugates.
About Roche
Headquartered in Basel, Switzerland, Roche is a leader in research-focused healthcare with combined strengths in pharmaceuticals and diagnostics. Roche is the world’s largest biotech company, with truly differentiated medicines in oncology, immunology, infectious diseases, ophthalmology and neuroscience. Roche is also the world leader in in vitro diagnostics and tissue-based cancer diagnostics, and a frontrunner in diabetes management. Roche’s personalised healthcare strategy aims at providing medicines and diagnostics that enable tangible improvements in the health, quality of life and survival of patients. Founded in 1896, Roche has been making important contributions to global health for more than a century. Twenty-nine medicines developed by Roche are included in the World Health Organization Model Lists of Essential Medicines, among them life-saving antibiotics, antimalarials and chemotherapy.
About AstraZeneca
AstraZeneca is a global, innovation-driven biopharmaceutical business that focuses on the discovery, development and commercialisation of prescription medicines, primarily for the treatment of cardiovascular, metabolic, respiratory, inflammation, autoimmune, oncology, infection and neuroscience diseases. AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. For more information please visit www.astrazeneca.com.
SEE………http://apisynthesisint.blogspot.in/2016/02/azd-9291-osimertinib-third-generation_4.html
/////
CN1C=C(C2=CC=CC=C21)C3=NC(=NC=C3)NC4=C(C=C(C(=C4)NC(=O)C=C)N(C)CCN(C)C)OC
Semagacestat (LY450139)

(2S)-2-hydroxy-3-methyl-N-((1S)-1-methyl-2-{[(1S)-3-methyl-2-oxo-2,3,4,5-tetrahydro-1H-3-benzazepin-1-yl]amino}-2-oxoethyl)butanamide
Semagacestat (LY450139) was a candidate drug
CEP-26401 Irdabisant; Histamine H3 Receptor Antagonists
1005402-19-6
313.3941, C18 H23 N3 O2
Histamine H3 Receptor Antagonists in phase 1
6-[4-[3-[2(R)-Methylpyrrolidin-1-yl]propoxy]phenyl]pyridazin-3(2H)-one
3(2H)-Pyridazinone, 6-[4-[3-[(2R)-2-methyl-1-pyrrolidinyl]propoxy]phenyl]-
6-(4-{3-[(2R)-2-methylpyrrolidin-1-yl]propoxy}phenyl)pyridazin-3(2H)-one
6-[4-[3-[(2R)-2-methyl-1-pyrrolidinyl]propoxy]phenyl]-3(2H)-pyridazinone
irdabisant
Cephalon Inc, innovator

CEP-26401 is a histamine H3 receptor antagonist in phase I clinical development at Cephalon to improve cognition in Alzheimer’s disease patients and for the treatment of schizophrenia. Cephalon was acquired by Teva in October 2011.
CEP-26401 [irdabisant; 6-{4-[3-((R)-2-methyl-pyrrolidin-1-yl)-propoxy]-phenyl}-2H-pyridazin-3-one HCl] is a novel, potent histamine H3 receptor (H3R) antagonist/inverse agonist with drug-like properties. High affinity of CEP-26401 for H3R was demonstrated in radioligand binding displacement assays in rat brain membranes (Ki = 2.7 ± 0.3 nM) and recombinant rat and human H3R-expressing systems (Ki = 7.2 ± 0.4 and 2.0 ± 1.0 nM, respectively).
CEP-26401 displayed potent antagonist and inverse agonist activities in [35S]guanosine 5′-O-(γ-thio)triphosphate binding assays. After oral dosing of CEP-26401, occupancy of H3R was estimated by the inhibition of ex vivo binding in rat cortical slices (OCC50 = 0.1 ± 0.003 mg/kg), and antagonism of the H3R agonist R-α-methylhistamine- induced drinking response in the rat dipsogenia model was demonstrated in a similar dose range (ED50 = 0.06 mg/kg).
CEP-26401 improved performance in the rat social recognition model of short-term memory at doses of 0.01 to 0.1 mg/kg p.o. and was wake-promoting at 3 to 30 mg/kg p.o. In DBA/2NCrl mice, CEP-26401 at 10 and 30 mg/kg i.p. increased prepulse inhibition (PPI), whereas the antipsychotic risperidone was effective at 0.3 and 1 mg/kg i.p. Coadministration of CEP-26401 and risperidone at subefficacious doses (3 and 0.1 mg/kg i.p., respectively) increased PPI. These results demonstrate potent behavioral effects of CEP-26401 in rodent models and suggest that this novel H3R antagonist may have therapeutic utility in the treatment of cognitive and attentional disorders.
CEP-26401 may also have therapeutic utility in treating schizophrenia or as adjunctive therapy to approved antipsychotics
. …………………………. str revealed by
By Carmen Drahl • Posted in http://cenblog.org/the-haystack/2011/03/drug-candidate-structures-revealed-at-acsanaheim/

……………………
WO 2008013838 or http://www.google.com/patents/EP2502918A1?cl=en
Example 11

Step 1.
-
[0174]

-
A mixture of 1-(4-hydroxyphenyl)ethanone (20.4 g, 150 mmol), K2CO3 (62.1 g, 3.0 eq.), and 3-bromo-1-chloropropane (29.6 mL, 2.0 eq.) in CH3COCH3 (200 mL) was heated to 65 °C overnight. The mixture was filtered, washed with acetone, and concentrated to dryness. The crude product was dissolved in 150 mL of CH2Cl2, and washed with saturated NaHCO3, NaCl solution and dried over Na2SO4. Concentration to dryness under vaccum afforded product (31.5 g, 99 % yield): MS m/z 213 (M + H).

Step 2.
-

-
A mixture of the product from step 1 1 (4.6 g, 1.0 eq.) and glyoxalic acid monohydrate (4.6g, 1.0 eq.) was stirred in 15 mL of acetic acid at 100 °C for 2 h. The solvent was evaporated and to the residue was added 25 mL of water, and cooled to 0 °C while conc. aqueous NH4OH was added to pH 8. To this mixture, hydrazine hydrate (4.76 mL, 2 eq.) was added and heated to 100 °C for 1 h. The resulting solid was filtered, washed with water. The crude material was dissolved in CH2Cl2/MeOH and purified by column chromatography with CH2Cl2 to 10 % MeOH in CH2Cl2; Mp 191-3 °C; MS m/z 265 (M + H).

Step 3.
-

-
A mixture of the product from step 2 (5.5 g, 21 mmol), K2CO3 (3.5 eq, 10.1g), 100 mg ofNaI, and R-2-methylpyrrolidine hydrochloride (2 eq., 5.1 g) in 250 mL of acetonitrile was heated to 80 °C for 2 days. The reaction mixture was then filtered, washed with CH2Cl2 (2 x 50mL), and concentrated. The residue was dissolved in 200 mL of CH2Cl2, and washed with saturated NaHCO3, saturated NaCl, dried with Na2SO4 and concentrated. The residue was purified by ISCO graduate chromatography with 100% CH2Cl2 to 5%MeOH: 95% CH2Cl2:0.5 mL of 2-aminopropane and then to 10%MeOH: 90% CH2Cl2:0.5 mL of 2-aminopropane to give the product. The product was dissolved in 15 mL of MeOH and then added 30 mL of 0.5 N HCl in EtOH. Evaporation of the solvent, and crystallization from MeOH: Et2O afforded the example 11 as the HCl salt (2.65g, 41 %): Mp 240-2 °C; MS m/z 314 (M + H).

……………….
Bioorg Med Chem Lett. 2012 Jun 15;22(12):4198-202. doi: 10.1016/j.bmcl.2012.04.001. http://www.sciencedirect.com/science/article/pii/S0960894X12004404
…………
Bioorganic and Medicinal Chemistry Letters, 2014 , vol. 24, 5 p. 1303 – 1306
http://www.sciencedirect.com/science/article/pii/S0960894X14000912

……………
Journal of Medicinal Chemistry, 2011 , vol. 54, 13 p. 4781 – 4792
http://pubs.acs.org/doi/full/10.1021/jm200401v

HTL-9936 is a selective muscarinic M1 agonist designed to improve cognitive function in patients with AD and other diseases

(2)

GENERAL STR
HTL-9936
PRE CLINICAL
Selective muscarinic acetylcholine receptor M1 (CHRM1; HM1) agonist
MolecularTargetMuscarinic acetylcholine receptor M1 (CHRM1) (HM1)
Mechanism of ActionMuscarinic acetylcholine receptor M1 agonist
Heptares Therapeutics was founded in 2007 to develop drugs against GPCRs. Its lead candidate,HTL-9936, is a selective muscarinic M1 agonist designed to improve cognitive function in patients with AD and other diseases, which recently entered the clinic for the first time.
Heptares Therapeutics, the leading GPCR structure-guided drug discovery and development company, announces that it has initiated a Phase 1 clinical study of HTL9936, the first fully selective muscarinic M1 receptor agonist to enter clinical development. HTL9936 is an orally available, small molecule drug candidate discovered using the Heptares GPCR structure-based drug design (SBDD) platform. Heptares plans to develop HTL9936 as a novel treatment for improving cognitive function (memory and thinking abilities) in patients with Alzheimer’s disease and other diseases associated with dementia and cognitive impairment.
“We are excited to initiate clinical development of HTL9936, a first-in-class agent with the potential to become an important new medicine for improving cognitive function in patients with Alzheimer’s disease and other potential indications including schizophrenia and Lewy body dementia,” said Malcolm Weir, CEO of Heptares. “In addition, the initiation of this clinical trial with HTL9936 marks an important milestone for Heptares, as we evolve into a clinical-stage business with a rich portfolio of novel GPCR-targeted agents advancing through Phase 1 and 2a clinical trials in the near-term.”
M1 receptor agonism is a well-validated mechanism of action for treating cognitive impairment and a valuable pharmacological profile that the pharmaceutical industry has endeavored to create for decades. The principal challenge has been to engineer selective compounds that activate the M1 receptor subtype without also activating the M2 or M3 receptors, which are associated with undesirable side effects. All previous compounds have been discontinued due to inadequate selectivity. Using a new structure-guided approach, Heptares scientists determined the x-ray crystal structure of the M1 receptor for the first time and leveraged unique insights into the receptor to identify new chemistries with fully selective M1 agonist profiles.
The Phase 1 study will evaluate the safety, tolerability and pharmacokinetics of HTL9936. In addition, the clinical pharmacodynamics of the drug will be investigated in a series of studies over the next year. This study aims to recruit more than 100 healthy volunteers including elderly people at a single clinical centre in the UK. Initial results are expected in mid-2014
About Alzheimer’s Disease and Other Disorders of Cognitive Impairment
Today there is significant unmet medical need and heavy economic burden across multiple diseases characterised by cognitive impairment and dementia. In Alzheimer’s disease, currently available drugs provide limited and transient effects on cognition. Healthcare costs associated with the epidemic of AD, including nursing home care, continue to grow dramatically and new therapies with better and more durable efficacy are urgently needed. In addition, an estimated 80% of schizophrenics suffer from cognitive impairment and 1.3 million patients in the US suffer from Lewy body dementia. Currently there are no approved therapies for treating cognitive impairment in schizophrenia or for treating Lewy body dementia.
About Heptares Therapeutics
Heptares creates new medicines targeting clinically important, yet historically challenging, GPCRs (G protein-coupled receptors), a superfamily of drug receptors linked to a wide range of human diseases. Leveraging our proprietary structure-based drug design technology platform, we have built an exciting pipeline of novel drug candidates with the potential to transform the treatment of serious diseases, including Alzheimer’s disease, ADHD, diabetes, schizophrenia, and migraine. Our pharmaceutical partners include Cubist, MorphoSys, Takeda, AstraZeneca and MedImmune, and we are backed by Clarus Ventures, MVM Life Science Partners, Novartis Venture Fund, the Stanley Family Foundation and Takeda Ventures. To learn more about Heptares, please visit http://www.heptares.com
…………………….
WO 2013072705
http://www.google.com/patents/WO2013072705A1?cl=en
Scheme 3 below.


Scheme 3
………………………………………….
WO 2014045031
http://www.google.com/patents/WO2014045031A1?cl=en
Muscarinic acetylcholine receptors (mAChRs) are members of the G protein-coupled receptor superfamily which mediate the actions of the neurotransmitter acetylcholine in both the central and peripheral nervous system. Five mAChR subtypes have been cloned, to M5. The mAChR is predominantly expressed post-synaptically in the cortex, hippocampus, striatum and thalamus; M2 mAChRs are located predominantly in the brainstem and thalamus, though also in the cortex, hippocampus and striatum where they reside on cholinergic synaptic terminals (Langmead et al., 2008 Br J
Pharmacol). However, M2 mAChRs are also expressed peripherally on cardiac tissue (where they mediate the vagal innervation of the heart) and in smooth muscle and exocrine glands. M3 mAChRs are expressed at relatively low level in the CNS but are widely expressed in smooth muscle and glandular tissues such as sweat and salivary glands (Langmead et al, 2008 Br J Pharmacol).
Muscarinic receptors in the central nervous system, especially the mAChR, play a critical role in mediating higher cognitive processing. Diseases associated with cognitive impairments, such as Alzheimer’s disease, are accompanied by loss of cholinergic neurons in the basal forebrain (Whitehouse et al, 1982 Science). In schizophrenia, which is also characterised by cognitive impairments, mAChR density is reduced in the pre-frontal cortex, hippocampus and caudate putamen of
schizophrenic subjects (Dean et al, 2002 Mol Psychiatry). Furthermore, in animal models, blockade or lesion of central cholinergic pathways results in profound cognitive deficits and non-selective mAChR antagonists have been shown to induce psychotomimetic effects in psychiatric patients. Cholinergic replacement therapy has largely been based on the use of acetylcholinesterase inhibitors to prevent the breakdown of endogenous acetylcholine. These compounds have shown efficacy versus symptomatic cognitive decline in the clinic, but give rise to dose-limiting side effects resulting from stimulation of peripheral M2 and M3mAChRs including disturbed gastrointestinal motility, bradycardia, nausea and vomiting
(http ://www. d rugs . com/pro/donepezi 1. htm I ;
http://yvww.drugs.com/pro/rivastigmine.html).
Scheme 1 below.
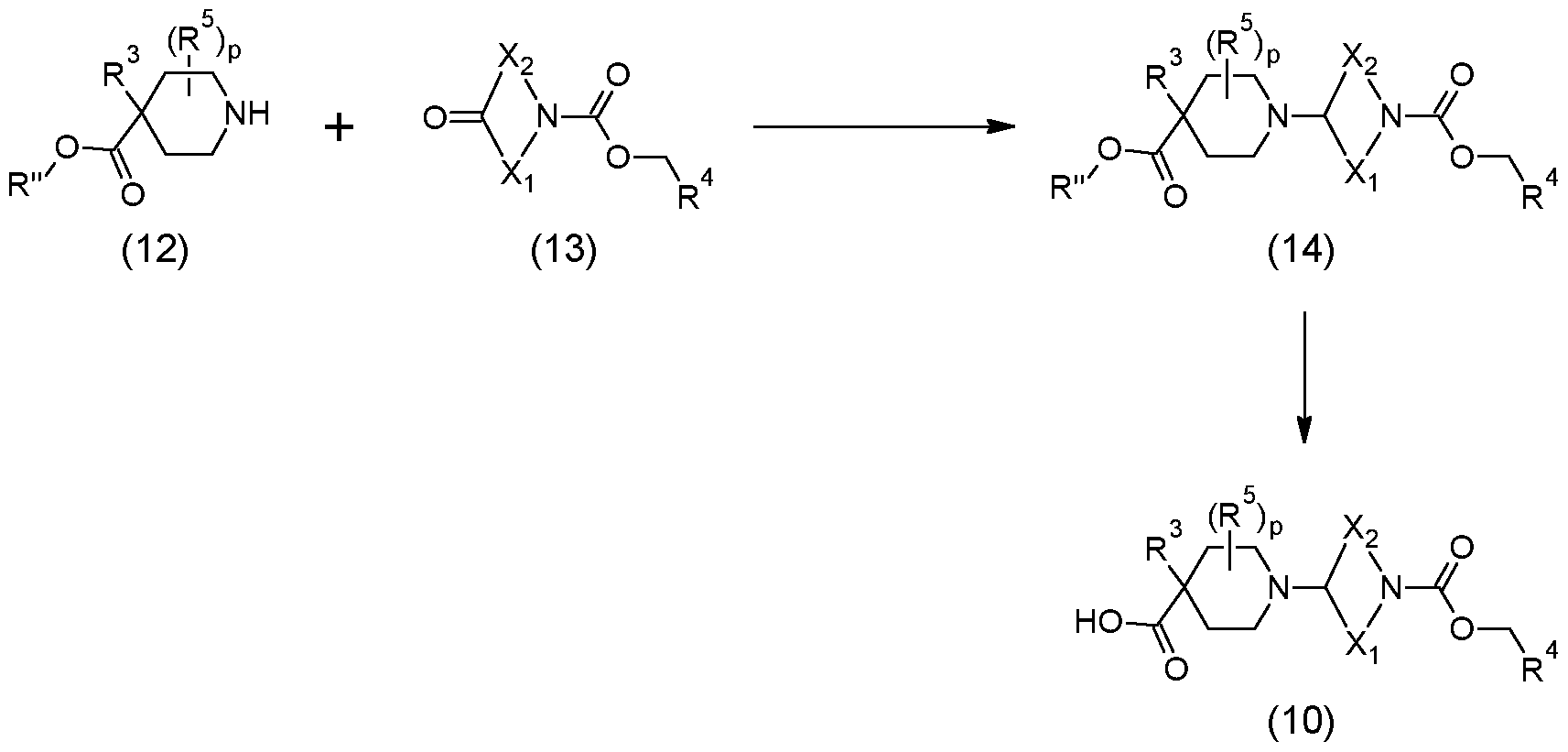
Scheme 1
Scheme 2 below.

(1 1)
Scheme 2
REF
March 2012, data were presented at the 243rd ACS meeting in San Diego, CA
April 2013, similar data were presented at the 245th ACS Meeting in New Orleans, LA.
September 2012, preclinical data were presented at the Fourth RSC/SCI GPCRs in Medicinal Chemistry Symposium in Windlesham, UK.
‘Achilles heel’ of pancreatic cancer identified
A research team at Georgetown Lombardi Comprehensive Cancer Center reports that inhibiting a single protein completely shuts down growth of pancreatic cancer, a highly lethal disease with no effective therapy.
Their study, published online today in Science Signaling, demonstrates in animal models and in human cancer cells that while suppressing Yes-associated protein (Yap) did not prevent pancreatic cancer from first developing, it stopped any further growth.
“We believe this is the true Achilles heel of pancreatic cancer, because knocking out Yap crushes this really aggressive cancer. This appears to be the critical switch that promotes cancer growth and progression,” says the study’s senior investigator, Chunling Yi, PhD, an assistant professor of oncology at Georgetown Lombardi.
Yi added that because Yap is over-expressed in other cancers, such as lung, liver and stomach tumors, researchers are already working on small molecule drugs that will inhibit activity of the protein and its…
View original post 229 more words
Kidney disease gene controls cancer highway
University of Queensland researchers have discovered that a gene that causes kidney disease also controls growth of the lymphatic system, a key route through which cancer spreads.
Pkd1 is the most frequently mutated gene in autosomal dominant polycystic kidney disease, which causes cysts to develop on kidneys and can lead to renal failure.
Researchers, led by Dr Ben Hogan from UQ’s Institute for Molecular Bioscience, (IMB) discovered that Pkd1 also controls lymphatic vessel development.
“Lymphatic vessels are used by tumours as a ‘highway’ through which they can metastasise, or spread, to other tissues,” Dr Hogan said.
“Most cancer deaths occur as a result of metastasis, so it is vital that we gain a better understanding of how lymphatic vessels grow and develop into a network.
“Pkd1 is a highly studied gene, so its unique role in lymphatic vessel formation is unexpected and gives us a unique entry point to…
View original post 120 more words

{kind=link}