
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Amgen Drug Evolocumab Hits Endpoint of Cholesterol Reduction
 Amgen announced that the Phase 3 TESLA (Trial Evaluating PCSK9 Antibody in Subjects with LDL Receptor Abnormalities) trial evaluating evolocumab met its primary endpoint of the percent reduction from baseline at week 12 in low-density lipoprotein cholesterol (LDL-C). The percent reduction in LDL-C, or “bad” cholesterol, was clinically meaningful and statistically significant………….read at
Amgen announced that the Phase 3 TESLA (Trial Evaluating PCSK9 Antibody in Subjects with LDL Receptor Abnormalities) trial evaluating evolocumab met its primary endpoint of the percent reduction from baseline at week 12 in low-density lipoprotein cholesterol (LDL-C). The percent reduction in LDL-C, or “bad” cholesterol, was clinically meaningful and statistically significant………….read at
| Monoclonal antibody | |
|---|---|
| Source | Human |
| Target | PCSK9 |
| Clinical data | |
| Legal status | ? |
| Identifiers | |
| CAS number | 1256937-27-5 |
| ATC code | None |
| Chemical data | |
| Formula | C6242H9648N1668O1996S56 |
| Mol. mass | 141.8 kDa |
Evolocumab[1] is a monoclonal antibody designed for the treatment of hyperlipidemia.[2] Evolocumab is a fully human monoclonal antibody that inhibits proprotein convertase subtilisin/kexin type 9 (PCSK9).

PCSK9 is a protein that targets LDL receptors for degradation and thereby reduces the liver’s ability to remove LDL-C, or “bad” cholesterol, from the blood.
Evolocumab, being developed by Amgen scientists, is designed to bind to PCSK9 and inhibit PCSK9 from binding to LDL receptors on the liver surface. In the absence of PCSK9, there are more LDL receptors on the surface of the liver to remove LDL-C from binding to LDL receptors on the liver surface. In the absence of PCSK9, there are more LDL receptors on the surface of the liver to remove LDL-C from the blood.

On 23 January 2014 Amgen announced that the Phase 3 GAUSS-2 (Goal Achievement After Utilizing an Anti-PCSK9 Antibody in Statin Intolerant Subjects-2) trial evaluating evolocumab in patients with high cholesterol who cannot tolerate statins met its co-primary endpoints: the percent reduction from baseline in low-density lipoprotein cholesterol (LDL-C) at week 12 and the mean percent reduction from baseline in LDL-C at weeks 10 and 12. The mean percent reductions in LDL-C, or “bad” cholesterol, compared to ezetimibe were consistent with results observed in the Phase 2 GAUSS study.[3]
The GAUSS-2 trial evaluated safety, tolerability and efficacy of evolocumab in 307 patients with high cholesterol who could not tolerate effective doses of at least two different statins due to muscle-related side effects. Patients were randomized to one of four treatment groups: subcutaneous evolocumab 140 mg every two weeks and oral placebo daily; subcutaneous evolocumab 420 mg monthly and oral placebo daily; subcutaneous placebo every two weeks and oral ezetimibe 10 mg daily; or subcutaneous placebo monthly and oral ezetimibe 10 mg daily.
Safety was generally balanced across treatment groups. The most common adverse events (> 5 percent in evolocumab combined group) were headache (7.8 percent evolocumab; 8.8 percent ezetimibe), myalgia (7.8 percent evolocumab; 17.6 percent ezetimibe), pain in extremity (6.8 percent evolocumab; 1.0 percent ezetimibe), and muscle spasms (6.3 percent evolocumab; 3.9 percent ezetimibe).

Evolocumab, a PCSK9 inhibitor, was safe and effective at lowering low-density lipoprotein cholesterol (LDL-C) after one year of treatment, according to a study published online Nov. 19 inCirculation and presented simultaneously at the American Heart Association scientific session in Dallas.
The Open-Label Study of Long-term Evaluation Against LDL-C (OSLER) trial took place at 156 study centers around the world that participated in at least one of four phase 2 studies of between October 2011 and June 2012. Evolocumab is a PCSK9 (proprotein convertase subtilisin/kexin type 9) inhibitor made by Amgen.
Investigators led by Michael J. Koren, MD, of the Jacksonville Center for Clinical Research in Florida, randomized 1,104 participants in a 2:1 ratio to receive either evolocumab (420 mg every four weeks) plus standard-of-care therapy (based on guidelines for treatment of hypercholesterolemia) or evolocumab alone, which served as the control. After 12 weeks, lipid results were unblinded and investigators were able to adjust standard-of-care therapy in either group.
The main efficacy objective was to determine the effects of longer-term evolocumab therapy on cholesterol levels and the main safety endpoints included incidence of adverse events, serious adverse events and adverse events resulting in discontinuation of the drug.
Patients who received evolocumab for the first time in the OSLER study had an average LDL-C reduction of 52.3 percent at one year. Patients previously dosed with evolocumab in a prior trial and were in the evolocumab and standard-of-care group in OSLER had an average LDL-C reduction of 52.1 percent at the end of the study compared with 50.4 percent at baseline. Patients who terminated evolocumab when they entered OSLER had their LDL-C levels returned to around their baseline.
Adverse events occurred in 73.1 percent of the standard-of-care group and 81.4 percent of the evolocumab plus standard-of-care group. The researchers determined that 5.6 percent of adverse events were related to evolocumab. Serious adverse events occurred in 6.3 percent of the control group and 7.1 percent in the combination group.
The authors explained that their findings offer more insight into the use of this class of drugs to lower LDL-C in at-risk patients.
“Challenging patients such as those who fail to reach current lipid goals despite maximum doses of highly effective statin agents or those with well-documented statin intolerance are thus logical populations for treatment with PCSK9 inhibitors,” they concluded.

References
- World Health Organization (2012). “International Nonproprietary Names for Pharmaceutical Substances (INN). Proposed INN: List 108” (PDF). WHO Drug Information 26 (4).
- Statement On A Nonproprietary Name Adopted By The USAN Council – Evolocumab
- Estel Grace Masangkay, “Amgen Phase III GAUSS-2 Trial of Evolocumab (AMG 145) Meets Co-Primary Endpoints Of LDL Cholesterol Reduction”, Bioresearch Online (January 24 2014)

NEW DRUG APPROVALS bY DR ANTHONY CRASTO WILL TOUCH 2 LAKH VIEWS THIS MONTH

NEW DRUG APPROVALS WILL TOUCH 2 LAKH VIEWS THIS MONTH
THANKS FOR YOUR SUPPORT
https://newdrugapprovals.org/ 2 LAKH VIEWS SOON
DR ANTHONY MELVIN CRASTO


MIFEPRISTONE

Mifepristone (or RU-486) is a synthetic steroid compound with both antiprogesterone and antiglucocorticoid properties. The compound is a 19-nor steroid with substitutions at positions C11 and C17 (17 beta-hydroxy-11 beta-[4-dimethylamino phenyl] 17 alpha-[1-propynyl]estra-4,9-dien-3-one), which antagonizes cortisol action competitively at the receptor level.
U.S. Pat. No. 4,386,085 (the ‘085 patent) discloses mifepristone starting from estra-5(10), 9(11)-diene-3,17-dione 3-ethylene acetal. The ‘085 patent discloses the purification of mifepristone by column chromatography using cyclohexane-ethyl acetate (7:3) mixture as an eluent. However, a drawback to the use of column chromatography is its unsuitability for industrial use.
Mifepristone is a progesterone receptor antagonist used as an abortifacient in the first months of pregnancy, and in smaller doses as an emergency contraceptive. Mifepristone is also a powerful glucocorticoid receptor antagonist, and has occasionally been used in refractory Cushing’s Syndrome(due to ectopic/neoplastic ACTH/Cortisol secretion). During early trials, it was known as RU-38486 or simply RU-486, its designation at the Roussel Uclaf company, which designed the drug. The drug was initially made available in France, and other countries then followed—often amid controversy. It is marketed under tradenames Korlym and Mifeprex, according to FDA Orange Book.
Mifepristone was the first antiprogestin to be developed and it has been evaluated extensively for its use as an abortifacient. The original target for the research group, however, was the discovery and development of compounds with antiglucocorticoid properties. It is these antiglucocorticoid properties that are of great interest in the treatment of severe mood disorders and psychosis.
In April 1980, as part of a formal research project at Roussel-Uclaf for the development of glucocorticoid receptorantagonists, chemist Georges Teutsch synthesized mifepristone (RU-38486, the 38,486th compound synthesized by Roussel-Uclaf from 1949 to 1980; shortened to RU-486); which was discovered to also be a progesterone receptor antagonist. In October 1981, endocrinologist Étienne-Émile Baulieu, a consultant to Roussel-Uclaf, arranged tests of its use for medical abortion in eleven women in Switzerland by gynecologist Walter Herrmann at theUniversity of Geneva‘s Cantonal Hospital, with successful results announced on April 19, 1982. On October 9, 1987, following worldwide clinical trials in 20,000 women of mifepristone with aprostaglandin analogue (initially sulprostone or gemeprost, later misoprostol) for medical abortion, Roussel-Uclaf sought approval in France for their use for medical abortion, with approval announced on September 23, 1988.
On October 21, 1988, in response to antiabortion protests and concerns of majority (54.5%) owner Hoechst AG of Germany, Roussel-Uclaf’s executives and board of directors voted 16 to 4 to stop distribution of mifepristone, which they announced on October 26, 1988. Two days later, the French government ordered Roussel-Uclaf to distribute mifepristone in the interests of public health.French Health Minister Claude Évin explained that: “I could not permit the abortion debate to deprive women of a product that represents medical progress. From the moment Government approval for the drug was granted, RU-486 became the moral property of women, not just the property of a drug company.” Following use by 34,000 women in France from April 1988 to February 1990 of mifepristone distributed free of charge, Roussel-Uclaf began selling Mifegyne (mifepristone) to hospitals in France in February 1990 at a price (negotiated with the French government) of $48 per 600 mg dose.
Mifegyne was subsequently approved in Great Britain on July 1, 1991, and in Sweden in September 1992, but until his retirement in late April 1994, Hoechst AG chairman Wolfgang Hilger, a devout Roman Catholic, blocked any further expansion in availability. On May 16, 1994, Roussel-Uclaf announced that it was donating without remuneration all rights for medical uses of mifepristone in the United States to the Population Council, which subsequently licensed mifepristone to Danco Laboratories, a new single-product company immune to antiabortion boycotts, which won FDA approval as Mifeprex on September 28, 2000.
On April 8, 1997, after buying the remaining 43.5% of Roussel-Uclaf stock in early 1997, Hoechst AG ($30 billion annual revenue) announced the end of its manufacture and sale of Mifegyne ($3.44 million annual revenue) and the transfer of all rights for medical uses of mifepristone outside of the United States to Exelgyn S.A., a new single-product company immune to antiabortion boycotts, whose CEO was former Roussel-Uclaf CEO Édouard Sakiz. In 1999, Exelgyn won approval of Mifegyne in 11 additional countries, and in 28 more countries over the following decade.
 mifepristone
mifepristone



The compound of structural formula 2 can be prepared from (+)-estrone in seven steps. Methylation of hydroxy group at C-3 in (+)-estrone, reduction of 17-ketone to 17β-alcohol followed by Birch reduction of ring A and mild hydrolysis of the enol ether to afford estra-17β-hydroxy-5(10)-en-3-one in four steps (Ref: Wilds, A. L. and Nelson, N. A. J. Am. Chem. Soc. 1953, 75, 5365-5369). This compound in another three steps, namely bromination and dehydrobrominatlon, ketalisation followed by Oppenauer oxidation yield compound having structural formula 2 (Ref: Perelman, M; Farkas, E.; Fornefield, E. J.; Kraay, R. J. and Rapala, B. T. J. Am. Chem. Soc. 1960, 82, 2402-2403).
U.S. Pat. No. 4,386,085 describes the synthesis of steroids of the general formula mentioned therein
-
In 50 cm3 of anhydrous tetrahydrofuran at 0, +5 ° C, bubbled up Allène the absorption of 2.1 g. Cooled to -70 ° C. and 15 minutes in 23.9 cm3 of a 1.3 M solution of butyllithium in hexanne. The resulting mixture is stirred for 15 minutes at -70 ° C.
- EXAMPLE 15 17β-hydroxy-11β-(4-dimethylaminophenyl)-17α (Propa-1 ,2-dienyl) estra-4 ,9-dien-3-one.Step A: 11β-(4-dimethylaminophenyl) 3,3 – / 1,2-ethane diyl bis (oxy) / 17α-(propa-1 ,2-dienyl) estr-9-en-5α-17β-diol and 11β – (4 – dimethylaminophenyl) 3,3 – / 1,2-ethane diyl bis (oxy) / 17α-(prop-2-ynyl) estr-9-en-5α (-17β-diol. Preparation of lithium compound.
Condensation
-
A solution of lithium derivative obtained above was added at -70 ° C in 25 minutes a solution of 3.5 g of the product obtained in Step A of Example 7 in 35 cm3 of anhydrous tetrahydrofuran. Stirred for 1 hour at -70 ° C, slowly poured into a saturated aqueous solution iced ammonium chloride. Extracted with ether, the organic phase washed with saturated sodium chloride, dried and the solvent evaporated. 3.4 g of product which was chromatographed on silica eluting with petroleum ether-ethyl acetate (1-1) to 1 mile triethylamine. Thus isolated: a) 1.73 g of isomer 17α-(propa-1 ,2-dienyl) F = 178 ° C. / Α / D = -32 ° ± 2 ° (c = 0.7% chloroform) b) 1.5 g of isomer 17o (- (prop-2-ynyl) F = 150 ° C. / α / D = -15 ° ± 2 ° (c = 0.9% chloroform).
Step B: 17β-hydroxy-11β-(4-dimethylaminophenyl)-17α (propa-1, 2 – dienyl) estra-4 ,9-dien-3-one.
-
Inert gas mixing 1.73 g of 17α isomer (- (propa-1, 2 – dienyl) obtained in Step A, 51.8 cm3 of 95% ethanol and 3.5 cm3 of 2N hydrochloric acid. stirred at 20 ° C for 1 hour, add 50 cm3 of methylene chloride and 50 cm3 of a 0.25 M solution of sodium bicarbonate, decanted, extracted with methylene chloride, washed with water, dried and the solvent evaporated. obtained 1.51 g of product was dissolved in 10 cm3 of methylene chloride hot. was added 15 cm3 of isopropyl ether, concentrated and allowed to stand. thus isolated 1.23 g of the expected product was crystallized again in methylene chloride-isopropyl ether. finally obtained 1.11 g of the expected product. F = 228 ° C.
/ Α / D – 139, 5 ° ± 3 ° (c = 0.8% chloroform). ANY ERROr MAIL ME amcrasto@gmail.com
 mifepristone
mifepristone|
5-27-1999
|
BORNEOL ESTERS, PROCESS FOR THEIR PREPARATION AND THEIR PHARMACEUTICAL USE
|
|
|
2-19-1999
|
NEW EPOTHILONE DERIVATIVES, METHOD FOR PRODUCING SAME AND THEIR PHARMACEUTICAL USE NEW EPOTHILONE DERIVATIVES, METHOD FOR PRODUCING SAME AND THEIR PHARMACEUTICAL USE
|
|
|
2-18-1999
|
Use of insect pheromones to treat pathologies induced by excess of glucocorticoid ANTIGLUCOCORTICOID DRUG
|
|
|
11-19-1998
|
BORNEOL DERIVATIVES AFFECTING TUBULIN POLYMERIZATION AND DEPOLYMERIZATION
|
|
|
10-3-1997
|
NOVEL BORNEOLS, PROCESSES FOR PRODUCING THEM AND PHARMACEUTICAL USE THEREOF
|
|
|
8-14-1997
|
11-Benzaldoximeestradiene-derivates, a process for their preparation and pharmaceutical compositions containing them
|
|
|
3-6-1997
|
New 11-benzaldehyde oxime, 17-beta methoxy, 17 alpha methoxymethyl derivates of estradiene, a process for their preparation and pharmaceutical compositions containing them
|
|
|
9-20-1995
|
Method for the diagnosis of cervical changes
|
|
|
12-22-1993
|
11-aryl steroid derivatives
|
|
8-25-2000
|
NOVEL EPOTHILONE DERIVATIVES, METHOD FOR PRODUCING THEM AND THEIR PHARMACEUTICAL USE NOVEL EPOTHILONE DERIVATIVES, METHOD FOR PRODUCING THEM AND THEIR PHARMACEUTICAL USE
|
|
|
8-25-2000
|
NOVEL EPOTHILON DERIVATIVES, METHOD FOR THE PRODUCTION THEREOF AND THEIR PHARMACEUTICAL APPLICATION NOVEL EPOTHILON DERIVATIVES, METHOD FOR THE PRODUCTION THEREOF AND THEIR PHARMACEUTICAL APPLICATION
|
|
|
8-25-2000
|
16-HALOGEN-EPOTHILONE DERIVATIVES, METHOD FOR PRODUCING THEM AND THEIR PHARMACEUTICAL USE 16-HALOGEN-EPOTHILONE DERIVATIVES, METHOD FOR PRODUCING THEM AND THEIR PHARMACEUTICAL USE
|
|
|
8-18-2000
|
EPOTHILON DERIVATIVES, METHOD FOR THE PRODUCTION AND THE USE THEREOF AS PHARMACEUTICALS EPOTHILON DERIVATIVES, METHOD FOR THE PRODUCTION AND THE USE THEREOF AS PHARMACEUTICALS
|
|
|
1-7-2000
|
EPOTHILON DERIVATIVES, THEIR PREPARATION PROCESS, INTERMEDIATE PRODUCTS AND THEIR PHARMACEUTICAL USE
|
|
|
12-17-1999
|
NOVEL ANTIESTROGENS, A METHOD FOR THE PRODUCTION THEREOF, AND THEIR PHARMACEUTICAL APPLICATION
|
|
|
11-26-1999
|
GLUCOCORTICOID RECEPTOR ANTAGONISTS FOR THE TREATMENT OF DEMENTIA
|
|
|
10-13-1999
|
Anti-glucocorticoid drug
|
|
|
9-30-1999
|
11 Beta-Benzaldoxim-9 Alpha, 10 Alpha-epoxy-estr-4-en-derivatives, a process for their production and pharmaceutical compositions containing them
|
|
|
9-11-1999
|
S-SUBSTITUTED 11 beta -BENZALDOXIME-ESTRA-4,9-DIENE-CARBONIC ACID THIOLESTERS, METHOD FOR THE PRODUCTION THEREOF AND PHARMACEUTICAL PREPARATIONS CONTAINING THESE COMPOUNDS
|
|
1-6-2011
|
PRODRUGS, THEIR PREPARATION AND USE AS MEDICAMENTS
|
|
|
6-4-2010
|
STEROID MODULATORS OF PROGESTERONE RECEPTOR AND/OR GLUCOCORTICOID RECEPTOR
|
|
|
7-26-2007
|
NEW EPOTHILONE DERIVATIVES, METHOD FOR PRODUCING SAME AND THEIR PHARMACEUTICAL USE
|
|
|
5-11-2007
|
Novel polymorph form M of mifepristone and process for its preparation
|
|
|
7-27-2006
|
GLYCOSYL PRODRUG CONJUGATE WITH ENHANCED TOLERANCE
|
|
|
7-13-2006
|
METHODS FOR TREATING PSYCHOSIS ASSOCIATED WITH GLUCOCORTICOID RELATED DYSFUNCTION
|
|
|
2-2-2006
|
Prodrugs for the treatment tumors and inflammatory diseases
|
|
|
2-7-2002
|
USE OF NITRIC OXIDE SYNTHASE SUBSTRATE AND/OR DONOR, OR A NITRIC OXIDE INHIBITOR FOR THE MANUFACTURE OF MEDICAMENTS FOR THE TREATMENT OF UTERINE CONTRACTILITY DISORDERS
|
|
|
9-13-2001
|
NOVEL BORNEOL DERIVATIVES, METHODS OF MANUFACTURING THEM, AND THEIR PHARMACEUTICAL USE
|
|
|
11-22-2000
|
Methods for treating psychosis associated with glucocorticoid related dysfunction
|

Nemonoxacin….TaiGen’s pneumonia antibiotic Taigexyn 奈诺沙星 gets marketing approval in Taiwan

Nemonoxacin 奈诺沙星
378746-64-6 CAS
TG-873870
-
C20-H25-N3-O4
- 371.4345
WARNER CHILCOTT ORIGINATOR
CLINICAL TRIALS http://clinicaltrials.gov/search/intervention=Nemonoxacin
(3S,5S)-7-[3-amino-5-methyl-piperidinyl]-l-cyclopropyl-l,4- dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid
7-[3(S)-Amino-5(S)-methylpiperidin-1-yl]-1-cyclopropyl-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid
Taigexyn has been approved in Taiwan IN 2014
13.03.14 |
TaiGen Biotechnology Receives Marketing Approval from the Taiwan Food and Drug Administration for Taigexyn in Taiwan
TAIPEI, March 13, 2014 /PRNewswire/ — TaiGen Biotechnology Company, Limited (“TaiGen”) today announced that the Taiwan Food and Drug Administration (TFDA) has approved the new drug application (NDA) of Taigexyn® (nemonoxacin) oral formulation (500 mg) for the treatment of community-acquired bacterial pneumonia (CAP). With this NDA approval, Taiwan is the first region to grant marketing approval to Taigexyn®. An NDA for Taigexyn® was also submitted to China FDA (CFDA) in April 2013 and is currently under review.
Nemonoxacin is a novel non-fluorinated quinolone antibiotic undergoing clinical trials.
Taigexyn Granted QIDP and Fast Track Designations
TaiGen Biotechnology announced that the FDA has granted nemonoxacin (Taigexyn) Qualified Infectious Disease Product (QIDP) and Fast Track designations for community-acquired bacterial pneumonia (CAP) and acute bacterial skin and skin structure infections (ABSSSI).
Nemonoxacin is a novel non-fluorinated quinolone broad spectrum antibiotic available in both oral and intravenous formulations. Nemonoxacin demonstrates activity against gram-positive and gram-negative bacteria and atypical pathogens. Nemonoxacin also possesses activities against methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant pathogens.
Nemonoxacin is a novel non-flourinated quinolone antibiotic registered in Taiwan for the oral treatment of community-acquired pneumonia. Clinical trials are in development at TaiGen Biotechnology for the treatment of diabetic foot infections and for the treatment of moderate to severe community-acquired pneumonia with an intravenous formulation. The drug is thought to accomplish its antibacterial action through topoisomerase inhibition.
Originally developed at Procter & Gamble, nemonoxacin was the subject of a strategic alliance formed in January 2005 between P&G and TaiGen to further the development and commercialization of nemonoxacin. In 2012, the product was licensed by TaiGen Biotechnology to Zhejiang Medicine in China for manufacturing, sales and marketing. In 2014, TaiGen out-licensed the exclusive rights of the product in Russian Federation, Commonwealth Independent States and Turkey to R-Pharm.
TaiGen has completed two Phase 2 clinical studies, one in CAP and the other in diabetic foot infections with demonstrated efficacy and safety. In the clinical trials conducted to date, nemonoxacin has shown activity against drug-resistant bacteria such as MRSA, quinolone-resistant MRSA, as well as quinolone-resistant Streptococcus pneumoniae.

Malate salt
Nemonoxacin malate anhydrous
951163-60-3 CAS NO, MW: 505.5209
Nemonoxacin malate hemihydrate
951313-26-1, MW: 1029.0566
Chemical structure of nemonoxacin as a malate salt (C20H25N3O4·C4H6O5·H2O). Nemonoxacin is the free base, and its molecular mass is 371.44 g/mol. The molecular mass of the salt, nemonoxacin malate, is 514.53 g/mol.


……………………..
isomeric compounds are:

(3S,5S)-7-[3-amino-5-methyl-piperidinyl]-l-cyclopropyl-l,4-dihydro-8- methoxy-4-oxo-3 -quinolinecarboxylic acid
COMPD1…….DESIRED

(3S,5R)-7-[3-amino-5-methyl-piperidinyl]-l-cyclopropyl-l,4-dihydro-8- methoxy-4-oxo-3 -quinolinecarboxylic acid
COMPD 1’….NOT DESIRED
Example 1
Malate salts of (3S,5S)-7-[3-amino-5-methyl-piperidinyl]-l-cyclopropyl-l,4- dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid (Compound 1) and (3S,5R)-7- [3-ammo-5-methyl-piperidinyl]- 1 -cyclopropyl- 1 ,4-dihydro-8-methoxy-4-oxo-3- quinolinecarboxylic acid (Compound 1′) were synthesized as follows:
(A) Synthesis of (3S,5S)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (Compound 9) and (3S,5R)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (Compound 9′): Compound 9′ was synthesized as shown in Scheme 1 below:
Scheme 1

3 4 Boc

A 50-L reactor was charged with Compound 2 (5.50 kg, 42.60 mol), methanol (27 L) and cooled to 10-150C. Thionyl chloride (10.11 kg, 2.0 equiv.) was added via an addition funnel over a period of 65 min, with external cooling to keep temperature below 30°. The resulting solution was stirred at 250C for 1.0 hour, after which methanol was removed under reduced pressure. The oily residue was azeotroped with ethyl acetate (3 x 2.5 L) to remove residual methanol, dissolved in ethyl acetate (27.4 L), charged into a 50 L reactor, and neutralized by slow addition of triethylamine (3.6 kg) below 3O0C. The resulting suspension was filtered to remove triethylamine hydrochloride.
The filtrate was charged to a 50 L reactor, along with DMAP (0.53 kg). Di- fert-butyl dicarbonate (8.43 kg) was added via hot water heated addition funnel, over a period of 30 min at a temperature of 20-300C. The reaction was complete after 1 hour as determined by TLC analysis. The organic phase was washed with ice cold IN HCl (2 x 7.5 L), saturated sodium bicarbonate solution (1 x 7.5 L), dried over magnesium sulfate, and filtered. After ethyl acetate was removed under reduced pressure, crystalline slurry was obtained, triturated with MTBE (10.0 L), and filtered to afford Compound 3 as a white solid (5.45 kg, 52.4%).
Anal. Calcd for CHHI7NO5 : C, 54.3; H, 7.04; N, 5.76. Found: C, 54.5; H, 6.96; N, 5.80. HRMS (ESI+) Expected for CHHI8NO5, [M+H] 244.1185. Found
244.1174; 1H NMR (CDCl3, 500 MHz):δ=4.54 (dd, J= 3.1, 9.5 Hz, IH), 3.7 (s, 3H), 2.58-2.50 (m, IH), 2.41 (ddd, IH, J= 17.6, 9.5, 3.7), 2.30-2.23 (m, IH), 1.98-1.93 (m, IH), 1.40 (s, 9H); 13C NMR (CDCl3, 125.70 MHz) δ 173.3, 171.9, 149.2, 83.5, 58.8, 52.5, 31.1, 27.9, 21.5. Mp 70.20C.
A 50-L reactor was charged with Compound 3 (7.25 kg, 28.8 mol), DME (6.31 kg), and Bredereck’s Reagent (7.7 kg, 44.2 mole). The solution was agitated and heated to 750C + 50C for three hours. The reaction was cooled to O0C over an hour, during which time a precipitate formed. The mixture was kept at O0C for an hour, filtered, and dried in a vacuum oven for at least 30 hours at 3O0C + 50C to give compound 4 as a white crystalline solid (6.93 kg, 77.9%).
Anal. Calcd for Ci4H22N2O5: C, 56.4; H, 7.43; N, 9.39. Found C, 56.4; H, 7.32; N, 9.48; HRMS (ESI+) Expected for Ci4H22N2O5, [M+H] 299.1607. Found 299.1613; 1H NMR (CDCl3, 499.8 MHz) δ = 7.11 (s, IH), 4.54 (dd, IH, J= 10.8, 3.6), 3.74 (s, 3H), 3.28-3.19 (m, IH), 3.00 (s, 6H), 2.97-2.85 (m,lH), 1.48 (s, 9H); 13C NMR (CDCl3, 125.7 MHz) δ = 172.6, 169.5, 150.5, 146.5, 90.8, 82.2, 56.0, 52.3, 42.0, 28.1, 26.3. MP 127.90C. A 10-gallon Pfaudler reactor was charged with ESCAT 142 (Engelhard Corp.
N.J, US) 5% palladium powder on carbon (50% wet, 0.58 kg wet wt), Compound 4 (1.89 kg, 6.33 mol), and isopropanol (22.4 Kg). After agitated under a 45-psi hydrogen atmosphere at 450C for 18 hrs, the reaction mixture was cooled to room temperature and filtered though a bed of Celite (0.51 kg). The filtrate was evaporated under reduced pressure to give a thick oil, which was solidified on standing to afford Compound 5 (1.69 kg, 100%) as a 93:7 diastereomeric mixture.
A sample of product mixture was purified by preparative HPLC to give material for analytical data. Anal. Calcd for Ci2Hi9NO5: C, 56.0; H, 7.44; N, 5.44. Found C, 55.8; H, 7.31; N, 5.44; MS (ESI+) Expected for Ci2Hi9NO5, [M+H] 258.1342. Found 258.1321; 1H NMR (CDCl3, 499.8 MHz) δ = 4.44 (m, IH), 3.72 (s, 3H), 2.60-2.48 (m, 2H), 1.59-1.54 (m, IH), 1.43 (s, 9H), 1.20 (d, j = 6.8 Hz,3H); 13C NMR (CDCl3, 125.7 MHz) δ = 175.7, 172.1, 149.5, 83.6, 57.4, 52.5, 37.5, 29.8, 27.9, 16.2. Mp 89.90C.
A 50-L reactor was charged with Compound 5 (3.02 kg, 11.7 mol), absolute ethanol (8.22 kg), and MTBE (14.81 kg). Sodium borohydride (1.36 kg, 35.9 mol) was added in small portions at 00C + 50C. A small amount of effervescence was observed. The reaction mixture was warmed to 1O0C + 50C and calcium chloride dihydrate (2.65 kg) was added in portions at 1O0C + 50C over an hour. The reaction was allowed to warm to 2O0C + 50C over one hour and agitated for an additional 12 hours at 200C + 50C. After the reaction was cooled to -50C + 50C, ice-cold 2N HCl (26.9 kg) was added slowly at of O0C + 50C. Agitation was stopped. The lower aqueous phase was removed. The reactor was charged with aqueous saturated sodium bicarbonate (15.6 kg) over five minutes under agitation. Agitation was stopped again and the lower aqueous phase was removed. The reactor was charged with magnesium sulfate (2.5 kg) and agitated for at leastlO minutes. The mixture was filtered though a nutsche filter, and concentrated under reduced pressure to afford Compound 6 (1.80 kg, 66%). Anal. Calcd for CnH23NO4: C, 56.6 H, 9.94; N, 6.00. Found C, 56.0; H, 9.68;
N, 5.96; HRMS (ESI+) Expected for CnH24NO4, [M+H] 234.1705. Found 234.1703; 1H NMR (CDCl3, 500 MHz) δ = 6.34 (d, J= 8.9 Hz, IH, NH), 4.51 (t, J= 5.8, 5.3 Hz, IH, NHCHCH2OH), 4.34 (t, J= 5.3, 5.3 Hz, IH, OBCHCH2OH), 3.46-3.45, (m, IH, NHCH), 3.28 (dd, J= 10.6, 5.3 Hz, NHCHCHHOH), 3.21 (dd, J= 10.2, 5.8 Hz , IH, CH3CHCHHOH), 3.16 (dd, J = 10.2, 6.2 Hz, IH, NHCHCHHOH), 3.12 (dd, J= 10.6, 7.1 Hz , IH, CH3CHCHHOH), 1.53-1.50 (m, IH, CH3CHCHHOH), 1.35 (s, 9H, 0(CHB)3, 1.30 (ddd, J = 13.9, 10.2, 3.7 Hz, IH, NHCHCHHCH), 1.14 (ddd, J= 13.6, 10.2, 3.4 Hz, IH, NHCHCHHCH), 0.80 (d, J= 6.6 Hz, 3H, CH3); 13C NMR (CDCl3, 125.7 MHz) δ 156.1, 77.9, 50.8, 65.1, 67.6, 65.1, 35.6, 32.8, 29.0, 17.1. Mp 92.10C. A 50 L reactor was charged with a solution of Compound 6 (5.1 kg) in isopropyl acetate (19.7 kg). The reaction was cooled to 150C + 5°C and triethylamine (7.8 kg) was added at that temperature. The reactor was further cooled to O0C + 50C and methanesulfonyl chloride (MsCl) (6.6 kg) was added. The reaction was stirred for a few hours and monitored for completion by HPLC or TLC. The reaction was quenched by saturated aqueous bicarbonate solution. The organic phase was isolated and washed successively with cold 10% aqueous triethylamine solution, cold aqueous HCl solution, cold saturated aqueous bicarbonate solution, and finally saturated aqueous brine solution. The organic phase was dried, filtered, and concentrated in vacuo below 550C + 50C to afford compound 7 as a solid/liquid slurry, which was used in the subsequent reaction without further purification.
After charged with 9.1 kg of neat benzylamine, a 50 L reactor was warmed to 550C, at which temperature, a solution of compound 7 (8.2 kg) in 1,2- dimethoxyethane (14.1 kg) was added. After the addition, the reaction was stirred at 6O0C + 50C for several hours and monitored for completion by TLC or HPLC. The reaction was cooled to ambient temperature and the solvent was removed under vacuum. The residue was diluted with 11.7 kg of 15% (v/v) ethyl acetate/hexanes solution and treated, while agitating, with 18.7 kg of 20% (wt) aqueous potassium carbonate solution. A triphasic mixture was obtained upon standing. The upper organic layer was collected. The isolated middle layer was extracted twice again with 11.7 kg portions of 15% (v/v) ethyl acetate/hexanes solution. The combined organic layers were concentrated under vacuum to give an oily residue. The residue was then purified by chromatography to afford Compound 8 as an oil. A 40 L pressure vessel was charged with 0.6 kg 50% wet, solid palladium on carbon (ElOl, 10 wt. %) under flow of nitrogen. A solution of Compound 8 (3.2 kg) in 13.7 kg of absolute ethanol was then added to the reactor under nitrogen. The reactor was purged with nitrogen and then pressurized with hydrogen at 45 psi. The reaction was then heated to 45°C. It was monitored by TLC or LC. Upon completion, the reaction was cooled to ambient temperature, vented, and purged with nitrogen. The mixture was filtered through a bed of Celite and the solid was washed with 2.8 kg of absolute ethanol. The filtrate was concentrated under vacuum to afford Compound 9 as a waxy solid.
TLC R/(Silica F254, 70:30 v/v ethyl acetate-hexanes, KMnO4 stain) = 0.12; 1H NMR (300 MHz, CDCl3) δ 5.31 (br s, IH), 3.80-3.68 (m, IH), 2.92 (d, J=I 1.4 Hz,
IH), 2.77 (AB quart, JAB=12.0 Hz, v=50.2 Hz, 2H), 2.19 (t, J=10.7 Hz, IH), 1.82-1.68 (m, 2H), 1.54 (br s, IH), 1.43 (s, 9H), 1.25-1.15 (m, IH), 0.83 (d, J=6.6 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ: 155.3, 78.9, 54.3, 50.8, 45.3, 37.9, 28.4, 27.1, 19.2; MS (ESI+) m/z 215 (M+H), 429 (2M+H). Similarly, (3S,5R)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester
(Compound 9′) was synthesized as shown in Scheme 2.
Scheme 2

HN Boc HN Boc
NaBH4,EtOH w – “ MsCI1TEA . „ _. – – _. „ Benzyl Amine
THF EA1CoId

(B) Synthesis of l-Cyclopropyl-7-fluoro-8-methoxy-4-oxo-l,4-dihydro-quinoline-3- carboxylic acid (Compound 10): Compound 10 was prepared according to the method described in U.S. Patent
6,329,391.
(C) Synthesis of borone ester chelate of l-Cyclopropyl-7-fluoro-8-methoxy-4-oxo- l,4-dihydro-quinoline-3-carboxylic acid (Compound 11):
Scheme 3

Toluene, tert-Butylmethyl ether 20-500C, filter
A reactor was charged with boron oxide (2.0 kg, 29 mol), glacial acetic acid (8.1 L, 142 mol), and acetic anhydride (16.2 L, 171 mol). The resulting mixture was refluxed at least 2 hours, and then cooled to 400C, at which temperature, 7- fluoroquinolone acid compound 10 (14.2 kg, 51 mol) was added. The mixture was refluxed for at least 6 hours, and then cooled to about 900C. Toluene (45 L) was added to the reaction. At 5O0C, terϊ-butylmethyl ether (19 L) was added to introduce precipitation. The mixture was then cooled to 200C and filtered to isolate the precipitation. The isolated solid was then washed with teτt-butylmethyl ether (26 L) prior to drying in a vacuum oven at 4O0C (50 torr) to afford Compound 11 in a yield of 86.4%. Raman (cm 1): 3084.7, 3022.3, 2930.8, 1709.2, 1620.8, 1548.5, 1468.0, 1397.7, 1368.3, 1338.5, 1201.5, 955.3, 653.9, 580.7, 552.8, 384.0, 305.8. NMR (CDCl3, 300 MHz) δ (ppm): 9.22 (s, IH), 8.38-8.33 (m, IH), 7.54 (t, J=9.8 Hz, IH), 4.38-4.35 (m, IH), 4.13 (s, 3H), 2.04 (s, 6H), 1.42-1.38 (m, 2H), 1.34-1.29 (m, 2H). TLC (Whatman MKC18F Silica, 6θA, 200 μm), Mobile Phase: 1 :1 (v/v) CH3CN : 0.5N NaCl (aq), UV (254/366 nm) visualization; R^O.4-0.5. (D) Synthesis of malate salt of (3S,5S)-7-[3-amino-5-methyl-piperidmyl]-l- cyclopropyl-l,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid (Compound 1) and malate salt of (3S,5R)-7-[3-amino-5-methyl-piperidmyl]-l-cyclopropyl-l,4- dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid (Compound 1′)
Compound 1 was synthesized from compound 9 as shown in Scheme 4 below:
Scheme 4

5O0C 3 d
a 6 0 N HCI (aq) CH2CI2 35°40°C 12 h t> Extract pH ad]ust to ~7-8 50″-65″C filter



A reactor was charged with Compound 11 (4.4 kg, 10.9 mol), Compound 9 (2.1 kg, 9.8 mol), triethylamine (TEA) (2.1 L, 14.8 mol), and acetonitrile (33.5 L, 15.7 L/kg). The resulting mixture was stirred at approximately 500C till completion of the reaction, as monitored by HPLC or reverse phase TLC. It was cooled to approximately 35°C and the reaction volume was reduced to approximately half by distillation of acetonitrile under vacuum between 0-400 torr. After 28.2 kg of 3.0 N NaOH (aq) solution was added, the reaction mixture was warmed to approximately 4O0C, distilled under vacuum until no further distillates were observed, and hydro lyzed at room temperature. Upon completion of hydrolysis, which was monitored by HPLC or reverse phase TLC, 4-5 kg of glacial acetic acid was added to neutralize the reaction mixture.
The resulting solution was extracted 3 times with 12.7 kg (9.6 L) of dichloromethane. The organic layers were combined and transferred to another reactor. The reaction volume was reduced to approximately a half by evaporation at 400C. After 20.2 Kg 6.0N HCl (aq) solution was added, the reaction mixture was stirred for at least 12 hours at 35°C. After the reaction was completed as monitored by HPLC or reverse phase TLC, agitation was discontinued to allow phase separation. The organic phase was removed and the aqueous layer was extracted with 12.7 kg (9.6 L) of dichloromethane. The aqueous layer was diluted with 18.3 kg distilled water and warmed to approximately 500C. Dichloromethane was further removed by distillation under vacuum (100-400 torr).
The pH of the aqueous solution was then adjusted to 7.8-8.1 by adding about 9.42 kg of 3.0 N NaOH (aq) below 65°C. The reaction mixture was stirred at 500C for at least an hour and then cooled to room temperature. The precipitate was isolated by suction filtration, washed twice with 5.2 kg of distilled water, and dried with suction for at least 12 hours and then in a convection oven at 55°C for additional 12 hours. Compound 12 (3.2 kg, 79%) was obtained as a solid.
A reactor was charged with 3.2 kg of Compound 12 and 25.6 kg of 95% ethanol. To the reactor was added 1.1 kg of solid D,L-malic acid. The mixture was refluxed temperature (~80°C). Distilled water (-5.7 L) was added to dissolve the precipice and 0.2 kg of activated charcoal was added. The reaction mixture was passed through a filter. The clear filtrate was cooled to 45°C and allowed to sit for at least 2 hours to allow crystallization. After the reaction mixture was further cooled to 5°C, the precipitate was isolated by suction filtration, washed with 6.6 kg of 95% ethanol, and dried with suction for at least 4 hours. The solid was further dried in a convection oven at 450C for at least 12 hours to afford 3.1 kg of Compound 1 (yield: 70%). NEMONOXACIN
NMR (D2O, 300 MHz) δ (ppm): 8.54 (s, IH), 7.37 (d, J=9.0 Hz, IH), 7.05 (d, J=9.0 Hz, IH), 4.23-4.18 (m, IH), 4.10-3.89 (m, IH), 3.66 (br s, IH), 3.58 (s, 3H), 3.45 (d, J=9.0 Hz, IH), 3.34 (d, J=9.3 Hz, IH), 3.16 (d, J=12.9 Hz, IH), 2.65 (dd, J=16.1, 4.1 Hz, IH), 2.64-2.53 (m, IH), 2.46 (dd, J=16.1, 8.0 Hz, IH), 2.06 (br s, IH), 1.87 (d, J=14.4 Hz, IH), 1.58-1.45 (m, IH), 1.15-0.95 (m, 2H), 0.91 (d, J=6.3 Hz, 3H), 0.85-0.78 (m, 2H).
Similarly, Compound 1′ was synthesized from Compound 9′ as shown in Scheme 5 below:
Scheme 5

(3S,5R)-7-[3-amino-5-methyl-piperidinyl]-l-cyclopropyl-l,4-dihydro-8- methoxy-4-oxo-3 -quinolinecarboxylic acid
COMPD 1’….NOT DESIRED
…………………
US2007/232650 A1,
malate salts of

(3S,5S)-7-[3-amino-5-methyl-piperidinyl]-1-cyclopropyl-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid (hereinafter Compound I, see also intermediate (23) in Section D, of Detailed Description of the Invention).
EXAMPLES Example 1 Synthesis of (3S,5S)-7-[3-amino-5-methyl-piperidinyl]-1-cyclopropyl-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid and malate salt thereof A. Synthesis of (3S,5S)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (8)

(2S)-1-(1,1-Dimethylethyl)-5-oxo-1,2-pyrrolidinedicarboxylic acid-2-methyl ester, (2). A 50-L reactor is charged with compound (1) (5.50 Kg, 42.60 mol), methanol (27 L) and cooled to 10-15° C. Thionyl chloride (10.11 Kg, 2.0 equiv.) is added via addition funnel over a period of 65 min, with external cooling to maintain temperature at <30°. The resulting solution is stirred at 25° C.+5° C. for 1.0 hour, after which the methanol is distilled off under reduced pressure. The resulting thick oil is azeotroped with ethyl acetate (3×2.5 L) to remove residual methanol. The residue is dissolved in ethyl acetate (27.4 L), charged into a 50 L reactor, and neutralized by the addition of triethylamine (3.6 Kg) from an addition funnel over 30 minutes. The temperature of the neutralization is maintained below 30° C. via external cooling. The resulting suspension of triethylamine hydrochloride is removed by filtration, and the clarified mother liquor solution is charged to a 50 L reactor, along with DMAP (0.53 Kg). Di-tert-butyl dicarbonate (8.43 Kg) is added via hot water heated addition funnel, over a period of 30 min with external cooling to maintain temperature at about 20-30° C. The reaction is complete after 1 hour as determined by TLC analysis. The organic phase is washed with ice cold 1N HCl (2×7.5 L), saturated sodium bicarbonate solution (1×7.5 L), and dried over magnesium sulfate. The mixture is filtered through a nutsche filter and ethyl acetate is removed under reduced pressure to yield a crystalline slurry that is triturated with MTBE (10.0 L) and filtered to afford intermediate (2) as a white solid (5.45 Kg, 52.4%). Anal. Calcd for C11H17NO5: C, 54.3; H, 7.04; N, 5.76. Found: C, 54.5; H, 6.96; N, 5.80. HRMS (ESI+) Expected for C11H18NO5, [M+H] 244.1185. Found 244.1174; 1H NMR (CDCl3, 500 MHz): δ=4.54 (dd, J=3.1, 9.5 Hz, 1H), 3.7 (s, 3H), 2.58-2.50 (m, 1H), 2.41 (ddd, 1H, J=17.6, 9.5, 3.7), 2.30-2.23 (m, 1H), 1.98-1.93 (m, 1H), 1.40 (s, 9H); 13C NMR (CDCl3, 125.70 MHz) δ 173.3, 171.9, 149.2, 83.5, 58.8, 52.5, 31.1, 27.9, 21.5; Mp 70.2° C.
(2S,4E)-1-(1,1-Dimethylethyl)-4-[(dimethylamino)methylene]-5-oxo-1,2-pyrrolidinedicarboxylic acid-2-methyl ester (3). A 50-L reactor is charged with intermediate (2) (7.25 Kg, 28.8 mol), DME (6.31 Kg), and Bredereck’s Reagent (7.7 Kg, 44.2 mole). The solution is agitated and heated to 75° C.±5° C. for at least three hours. The progress of the reaction is monitored by HPLC. The reaction is cooled to 0° C.±5° C. over on hour during which time a precipitate forms. The mixture is held at 0° C.±5° C. for one hour and filtered though a nutsche filter and the product dried in a vacuum oven for at least 30 hours at 30° C.±5° C. to give intermediate (3) as a white crystalline solid (6.93 Kg, 77.9%). Anal. Calcd for C14H22N2O5: C, 56.4; H, 7.43; N, 9.39. Found C, 56.4; H, 7.32; N, 9.48; HRMS (ESI+) Expected for C14H22N2O5, [M+H] 299.1607. Found 299.1613; 1H NMR(CDCl3, 499.8 MHz)δ=7.11 (s, 1H), 4.54 (dd, 1H, J=10.8, 3.6), 3.74 (s, 3H), 3.28-3.19 (m, 1H), 3.00 (s, 6H), 2.97-2.85 (m, 1H), 1.48 (s, 9H); 13C NMR (CDCl3, 125.7 MHz) δ=172.6, 169.5, 150.5, 146.5, 90.8, 82.2, 56.0, 52.3, 42.0, 28.1, 26.3. Mp 127.9° C.
(2S,4S)-1-(1,1-Dimethylethyl)-4-methyl-5-oxo-1,2-pyrrolidinedicarboxylic acid-2-methyl ester (4). A 10-gallon Pfaudler reactor is inerted with nitrogen and charged with ESCAT 142 5% palladium powder on carbon (50% wet, 0.58 Kg wet wt.), intermediate (3) (1.89 Kg, 6.33 mol) and isopropanol (22.4 Kg). The reaction mixture is agitated under a 45-psi hydrogen atmosphere at 45° C. for 18 hrs. The reaction mixture is then cooled to room temperature and filtered though a bed of Celite (0.51 Kg) in a nutsche filter to remove catalyst. The mother liquor is evaporated under reduced pressure to give a thick oil that crystallizes on standing to afford 4 (1.69 Kg, 100%) as a 93:7 diastereomeric mixture. A sample of product mixture is purified by preparative HPLC to give material for analytical data. Anal. Calcd for C12H19NO5: C, 56.0; H, 7.44; N, 5.44. Found C, 55.8; H, 7.31; N, 5.44; MS (ESI+) Expected for C12H19NO5, [M+H] 258.1342. Found 258.1321; 1H NMR (CDCl3, 499.8 MHz) δ=4.44 (m, 1H), 3.72 (s, 3H), 2.60-2.48 (m, 2H), 1.59-1.54 (m, 1H), 1.43 (s, 9H), 1.20 (d, j=6.8 Hz,3H); 13C NMR (CDCl3, 125.7 MHz) δ=175.7, 172.1, 149.5, 83.6, 57.4, 52.5, 37.5, 29.8, 27.9, 16.2. Mp 89.9° C.
(1S,3S)-(4-Hydroxyl-1-hydroxymethyl-3-methyl-butyl)-carbamic acid tert-butyl ester (5). A 50-L reactor is charged with intermediate (4) (3.02 Kg, 11.7 mol), absolute ethanol (8.22 Kg), and MTBE (14.81 Kg). The solution is agitated and cooled to 0° C.±5° C. and sodium borohydride (1.36 Kg, 35.9 mol) is added in small portions so as to maintain reaction temperature at 0° C.±5° C. A small amount of effervescence is observed. The reaction mixture is warmed to 10° C.±5° C. and calcium chloride dihydrate (2.65 Kg) is added portion wise at a slow rate over an hour so as to maintain a reaction temperature of 10° C.±5° C. The reaction is allowed to warm to 20° C.±5° C. over one hour and agitated for an additional 12 hours at 20° C.±5° C. The reaction is cooled to −5° C.±5° C., ice-cold 2N HCl (26.9 Kg) is added at a rate to maintain a reaction temperature of 0° C.±5° C. Agitation is stopped to allow phases to separate. The lower aqueous phase (pH=1) is removed. The reactor is charged with aqueous saturated sodium bicarbonate (15.6 Kg) over five minutes. Agitation is stopped to allow phases to separate. The lower aqueous phase (pH=8) is removed. The reactor is charged with magnesium sulfate (2.5 Kg) and agitated for at least 10 minutes. The mixture is filtered though a nutsche filter, and condensed under reduced pressure to afford intermediate (5) (1.80 Kg, 66%). Anal. Calcd for C11H23NO4: C, 56.6; H, 9.94; N, 6.00. Found C, 56.0; H, 9.68; N, 5.96; HRMS (ESI+) Expected for C11H24NO4, [M+H] 234.1705. Found 234.1703; 1H NMR (CDCl3, 500 MHz)δ=6.34(d, J=8.9 Hz, 1H, NH), 4.51 (t, J=5.8, 5.3 Hz, 1H, NHCHCH2OH), 4.34 (t, J=5.3, 5.3 Hz, 1H, CH3CHCH2OH), 3.46-3.45, (m, 1H, NHCH), 3.28 (dd, J=10.6, 5.3 Hz, NHCHCHHOH), 3.21 (dd, J=10.2, 5.8 Hz, 1H, CH3CHCHHOH), 3.16 (dd, J=10.2, 6.2 Hz, 1H, NHCHCHHOH), 3.12 (dd, J=10.6, 7.1 Hz, 1H, CH3CHCHHOH), 1.53-1.50 (m, 1H, CH3CHCHHOH), 1.35 (s, 9H, O(CH 3)3, 1.30 (ddd, J=13.9, 10.2, 3.7 Hz, 1H, NHCHCHHCH), 1.14 (ddd, J=13.6, 10.2, 3.4 Hz, 1H, NHCHCHHCH), 0.80 (d, J=6.6 Hz, 3H, CH3); 13C NMR (CDCl3, 125.7 MHz) δ 156.1, 77.9, 50.8, 65.1, 67.6, 65.1, 35.6, 32.8, 29.0, 17.1. Mp 92.1° C.
(2S,4S)-Methanesulfonic acid 2-tert-butoxycarbonylamino-5-methanesulfonyloxy-4-methyl-pentyl ester (6). A 50 L reactor is charged with a solution of intermediate (5) (5.1 Kg) in isopropyl acetate (i-PrOAc) 11.8 Kg followed by a rinse with an additional 7.9 Kg i-PrOAc. The reaction is cooled to 15° C.±5° C. and triethylamine (TEA) (7.8 Kg) is added while maintaining the set temperature. The reactor is further cooled to 0° C.±5° C. and methanesulfonyl chloride (MsCl) (6.6 Kg) is added to the reaction solution while maintaining the set temperature. The reaction is stirred for a few hours and monitored for completion by HPLC or TLC. The reaction is quenched by the addition of a saturated aqueous bicarbonate solution and the resulting isolated organic phase is washed successively with cold 10% aqueous triethylamine solution, cold aqueous HCl solution, cold saturated aqueous bicarbonate solution, and finally saturated aqueous brine solution. The organic phase is dried, filtered, and concentrated in vacuo below 55° C.±5° C. until a solid/liquid slurry containing intermediate (6) is obtained. The slurry is used crude in subsequent reaction without further characterization.
(3S,5S)-(1-Benzyl-5-methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (7). A 50 L reactor is charged with 9.1 Kg of neat benzylamine. The reactor is brought to 55° C. and a solution of intermediate (6) (8.2 Kg) in 1,2-dimethoxyethane (DME) (14.1 Kg) is added to the reactor while maintaining a temperature of 60° C.±5° C. After complete addition of this solution, the reaction is stirred at 60° C.±5° C. for several hours and monitored for completion by TLC or HPLC. The reaction is cooled to ambient temperature and volatiles (DME) are removed by rotary evaporation under vacuum. The residue is diluted with 11.7 Kg of 15% (v/v) ethyl acetate/hexanes solution and treated, while agitating, with 18.7 Kg of 20% (wt) aqueous potassium carbonate solution. A triphasic mixture is obtained upon settling. The bottom aqueous phase is removed and the middle phase is set aside. The upper organic phase is collected and held for combination with extracts from additional extractions. The isolated middle phase is extracted twice again with 11.7 Kg portions of 15% (v/v) ethyl acetate/hexanes solution, each time combining the extracts with original organic phase. The combined organic extracts are transferred into a rotary evaporator and solvent is removed under vacuum until an oily residue remains. The residue is then purified via large-scale preparative chromatography to afford purified intermediate (7) as an oil.
(3S,5S)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (8). A 40 L pressure vessel is charged with 0.6 Kg 50% wet, solid palladium on carbon (E101, 10 wt. %) under flow of nitrogen. A solution of 3.2 Kg intermediate (7) in 13.7 Kg of absolute ethanol is then charged to the reactor under nitrogen. The reactor is purged with nitrogen and is then pressurized with hydrogen at 45 psi. The reaction is then heated to 45° C. while maintaining a hydrogen pressure of 45 psi. The reaction is monitored by TLC or LC until complete. The reaction is cooled to ambient temperature, vented, and purged with nitrogen. The reactor contents are filtered through a bed of Celite and the solids are washed with 2.8 Kg of absolute ethanol. The filtrate is concentrated by rotary evaporation under vacuum until a waxy solid is obtained to afford intermediate (8): TLC Rf (Silica F254, 70:30 v/v ethyl acetate-hexanes, KMnO4 stain)=0.12; 1H NMR (300 MHz, CDCl3) δ 5.31 (br s, 1H), 3.80-3.68 (m, 1H), 2.92 (d, J=11.4 Hz, 1H), 2.77 (AB quart, JAB=12.0 Hz, Δν=50.2 Hz, 2H), 2.19 (t, J=10.7 Hz, 1H), 1.82-1.68 (m, 2H), 1.54 (br s, 1H), 1.43 (s, 9H), 1.25-1.15 (m, 1H), 0.83 (d, J=6.6 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 155.3, 78.9, 54.3, 50.8, 45.3, 37.9, 28.4, 27.1, 19.2; MS (ESI+) m/z 215 (M+H), 429 (2M+H).
B. Synthesis of 1-Cyclopropyl-7-fluoro-8-methoxy-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (19)


Intermediate (12): A reactor is charged with a solution of intermediate (11) (1.2 Kg, 7.7 mol, 1.0 eq) in anhydrous toluene (12 L) followed by ethylene glycol (1.8 L, 15.7 mol, 4.2 eq) and solid p-toluenesulfonic acid (120 g, 10 wt. %). The reaction mixture is stirred at ambient temperature for at least 30 minutes and then heated to reflux, collecting the water/toluene azeotrope in a Dean Stark type trap apparatus until the reaction is complete as determined by TLC analysis (15% EtOAc/Hexanes v/v). Upon completion, the reaction is cooled to ambient temperature and poured into an aqueous solution of sodium bicarbonate (6 L). The organic toluene phase was removed and washed with saturated sodium bicarbonate solution (6 L), distilled water (2×6 L), and saturated aqueous brine (6 L). The organic phase was removed and dried over MgSO4, filtered, and evaporated under reduced pressure to afford intermediate (12) as an oil (1.3 Kg, 86%). The material is used without further purification in subsequent reaction steps.
Intermediate (13): A reactor is charged with a solution of intermediate (12) (1.2 Kg, 6.0 mol, 1.0 eq) in anhydrous tetrahydrofuran (12 L) and n-butyllithium (2.5M in hexanes, 2.6 L, 6.6 mol, 1.1 eq) is added at −40° C., while maintaining this temperature throughout the addition. The reaction is stirred for at least one hour at −40° C. and trimethylborate (0.9 L, 7.8 mol, 1.3 eq) is added to the mixture while maintaining the temperature at or below −40° C. The reaction mixture is stirred for at least one hour at −40° C. until complete as determined by TLC analysis (30% EtOAc/Hexanes v/v). The reaction is warmed slightly to −30° C. and acetic acid (3 L) is added slowly. Upon complete addition, water is added (0.5 L) to the reaction and the mixture is allowed to quickly warm to ambient temperature while stirring overnight. Organic solvent is removed from the reaction by distillation under reduced pressure at 45° C. To the reaction residue is added 3-4 volumes of water (6 L) and 30% hydrogen peroxide (0.7 L, 1.0 eq) slowly at ambient temperature with cooling provided to control the exotherm. The reaction is stirred for at least an hour at ambient temperature until complete as determined by TLC (15% EtOAc/Hexanes v/v). The reaction mixture is cooled to 0-5° C. and excess peroxide is quenched with the addition of 10% aqueous sodium bisulfite solution (2 L). The mixture is tested to ensure a negative peroxide result and the reaction is acidified by the addition of 6N HCl (aq) (1.2 L). The reaction is stirred until the hydrolysis reaction is complete as determined by TLC or NMR analysis. The resulting solids are collected by suction filtration to afford intermediate (13) as a yellow solid (1.0 Kg, 79%).
Intermediate (14): A reactor is charged with intermediate (13) (0.53 Kg, 3.0 mol, 1.0 eq) and dissolved in dry toluene (2.7 Kg, 3.1 L). To this solution is added dimethylsulfate (0.49 Kg, 3.9 mol, 1.30 eq) followed by solid potassium carbonate (0.58 Kg, 4.2 mol, 1.4 eq). The reaction mixture is heated to reflux and held for at least 1 hour until complete as determined by HPLC. During this time, vigorous gas evolution is observed. The reaction is then cooled to ambient temperature and diluted with distilled water (3.2 L) along with 30% NaOH (aq) (0.13 Kg, 0.33 eq). The aqueous phase is separated and the remaining toluene phase is extracted twice more with distilled water (3.2 L) combined with 30% NaOH (aq) (0.13 Kg, 0.33 eq), removing the aqueous phase each time. The organic upper phase is concentrated by distillation in vacuo (<100 mbar) at approximately 40° C. until a concentrated toluene solution is achieved. The resulting solution is cooled to ambient temperature, checked for quality and yield by HPLC, and carried forward to the next step in the synthesis without further purification (theoretical yield for intermediate (14) assumed, 0.56 Kg).
Intermediate (15a,b): A reactor is charged with 1.8 Kg (2.1 L) anhydrous toluene along with sodium hydride (0.26 Kg, 6.6 mol, 2.20 eq) as a 60 wt. % dispersion in mineral oil. To this mixture is added (0.85 Kg, 7.2 mol, 2.4 eq) diethylcarbonate as the reaction mixture is heated to 90° C. over 1 hour. A solution of intermediate (14) (˜1.0 eq) in toluene from the previous step is added to the reaction while maintaining a temperature of 90° C.±5° C. Gas evolution can be observed during this addition. After complete addition, the reaction is stirred for at least 30 minutes or until complete as determined by HPLC analysis. Upon completion, the mixture is cooled to ambient temperature and diluted with 10 wt. % aqueous sulfuric acid (3.8 Kg, 3.9 mol, 1.3 eq) with agitation. The phases are allowed to separate and the lower aqueous phase is removed. The remaining organic phase is concentrated in vacuo (<100 mbar) at approximately 40° C. until a concentrated toluene solution is achieved. The resulting solution is cooled to ambient temperature and carried forward to the next step in the synthesis without further purification (theoretical yield for intermediate (15a,b) assumed, 0.85 Kg).
Intermediate (16a,b; 17a,b): A reactor is charged with a solution of intermediate (15a,b) (0.85 Kg, ˜3.0 mol, ˜1.0 eq) in toluene from the previous step. To the reactor is then added dimethylformamide-dimethylacetal (0.54 Kg, 4.5 mol, 1.5 eq) and the resulting solution is heated to reflux temperature (˜95-105° C.). The lower boiling solvent (methanol from reaction) is allowed to distill off while the temperature is maintained at ≧90° C. Heating is continued for at least 1 hour or until complete as determined by HPLC analysis. Upon completion, the reaction containing the mixture of intermediate (16a,b), is cooled to ambient temperature and toluene (1.8 Kg, 2.1 L) along with cyclopropylamine (0.21 Kg, 3.6 mol, 1.2 eq) are added to the reaction. The reaction is stirred at ambient temperature for at least 30 minutes until complete as determined by HPLC. Upon completion, the reaction is diluted with 10 wt. % aqueous sulfuric acid (2.9 Kg, 3.0 mol, 1.0 eq) with agitation, and the phases are then allowed to separate. The aqueous phase is removed and the organic phase is concentrated under reduced pressure (<100 mbar) at approximately 40° C. by distillation. When the desired concentration is achieved, the solution is cooled to ambient temperature and the toluene solution containing the mixture of intermediate (17a,b) is carried forward to the next step in the synthesis without further purification (theoretical yield for intermediate (17a,b) assumed, ˜1.1 Kg).
Intermediate (18): A reactor is charged with a solution of the mixture of intermediate (17a,b) (˜4.7 Kg, ˜3.0 mol) at ambient temperature. To the reactor is added N,O-bis(trimethylsilyl)acetamide (0.61 Kg, 3.0 mol, 1.0 eq) and the reaction is heated to reflux temperature (˜105-115° C.) for at least 30 minutes or until complete as determined by HPLC analysis. If not complete, an additional amount of N,O-bis(trimethylsilyl)acetamide (0.18 Kg, 0.9 mol, 0.3 eq) is added to the reaction to achieve completion. Upon completion, the reaction is cooled to below 40° C. and organic solvent is removed under reduced pressure (<100 mbar) at approximately 40° C. by distillation until a precipitate is formed. The reaction is cooled to ambient temperature and the precipitated solids are isolated by suction filtration and washed with distilled water twice (1×1.8 L, 1×0.9 L). The solid is dried to afford intermediate (18) as a white solid (0.76 Kg, 82%). The material is used without further purification in the next reaction step.
Intermediate (19): A reactor is charged with solid intermediate (18) (0.76 Kg, ˜2.5 mol, ˜1.0 eq) at ambient temperature followed by ethanol (5.3 Kg, 6.8 L) and 32 wt. % aqueous hydrochloric acid (1.1 Kg, 10 mol). The reaction mixture is brought to reflux temperature (76-80° C.) during which time the mixture first becomes homogeneous and later becomes heterogeneous. The mixture is heated at reflux for at least 5 hours or until complete as determined by TLC analysis (15% EtOAc/Hexanes v/v). Upon completion, the reaction is cooled to 0° C.±5° C. and the precipitated solid is isolated by filtration and washed with distilled water (1.7 Kg) followed by ethanol (1.7 Kg). The isolated solid is dried to afford intermediate (19) as a white solid (0.65 Kg, ˜95%). 1H NMR (CDCl3, 300 MHz) δ (ppm): 14.58 (s, 1H), 8.9 (s, 1H), 8.25 (m, 1H), 7.35 (m, 1H), 4.35 (m, 1H), 4.08 (s, 3H), 1.3 (m, 2H), 1.1 (m, 2H) 19F NMR (CDCl3+CFCl3, 292 MHz) δ (ppm): −119. HPLC: 99.5% by area.
C. Synthesis of borone ester chelate of 1-Cyclopropyl-7-fluoro-8-methoxy-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (20)

A reactor is charged with boron oxide (2.0 Kg, 29 mol) followed by dilution with glacial acetic acid (8.1 L, 142 mol) and acetic anhydride (16.2 L, 171 mol). The resulting mixture is heated to reflux temperature for at least 2 hours. The reaction contents are cooled to 40° C. and the solid 7-fluoroquinolone acid intermediate (19) (14.2 Kg, 51 mol) is added to the reaction mixture. The mixture is again heated to reflux temperature for at least 6 hours. Reaction progress is monitored by HPLC and NMR. The mixture is cooled to approximately 90° C. and toluene (45 L) is added to the reaction. The reaction is further cooled to 50° C. and tert-butylmethyl ether (19 L) is added to the reaction mixture to bring about precipitation of the product. The mixture is then cooled to 20° C. and the solid product 19 is isolated by filtration. The isolated solids are then washed with tert-butylmethyl ether (26 L) prior to drying in a vacuum oven at 40° C. (50 torr). The product yield obtained for intermediate (20) in this reaction is 86.4%. Raman (cm−1): 3084.7, 3022.3, 2930.8, 1709.2, 1620.8, 1548.5, 1468.0, 1397.7, 1368.3, 1338.5, 1201.5, 955.3, 653.9, 580.7, 552.8, 384.0, 305.8. NMR (CDCl3, 300 MHz) δ (ppm): 9.22 (s, 1H), 8.38-8.33 (m, 1H), 7.54 (t, J=9.8 Hz, 1H), 4.38-4.35 (m, 1H), 4.13 (s, 3H), 2.04 (s, 6H), 1.42-1.38 (m, 2H), 1.34-1.29 (m, 2H). TLC (Whatman MKC18F Silica, 60 Å, 200 μm), Mobile Phase: 1:1 (v/v) CH3CN:0.5N NaCl (aq), UV (254/366 nm) visualization; Rf=0.4-0.5.
D. Coupling of 1-Cyclopropyl-7-fluoro-8-methoxy-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (20) to (3S,5S)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (8), and synthesis of malate salt of (3S,5S)-7-[3-amino-5-methyl-piperidinyl]-1-cyclopropyl-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid (25)

A reactor is charged with solid intermediate (20) (4.4 Kg, 10.9 mol) followed by dilution with a solution of triethylamine (TEA) (2.1 L, 14.8 mol) and piperidine side chain intermediate (8) (2.1 Kg, 9.8 mol) in acetonitrile (33.5 L, 15.7 L/Kg) at room temperature. The resulting mixture is warmed to approximately 50° C. until reaction is judged complete. Reaction progress is monitored by HPLC or reverse phase TLC. When complete, the reaction is cooled to approximately 35° C. and reaction volume is reduced to approximately half by distillation of acetonitrile under vacuum between 0-400 torr. The reactor is then charged with 28.2 Kg of 3.0N NaOH (aq) solution and the temperature is raised to approximately 40° C. Distillation under vacuum is continued between 1-4 hours or until no further distillates are observed. The reaction is then cooled to room temperature and the hydrolysis reaction is monitored by HPLC or reverse phase TLC. Upon completion, the reaction mixture is neutralized to a pH of between 6-8 by adding ˜4-5 Kg of glacial acetic acid. The reactor is then charged with 12.7 Kg (9.6 L) of dichloromethane as an extraction solvent, the mixture is agitated, phases are allowed to separate, and the organic dichloromethane phase is removed. The extraction process is repeated two additional times using 12.7 Kg (9.6 L) of dichloromethane, collecting the lower, organic phase each time. The aqueous phase is discarded and the organic extracts are combined in a single reactor. The reactor contents are heated to 40° C. and the reaction volume is reduced to approximately one half by distillation. The reactor is then charged with 20.2 Kg 6.0N HCl (aq) solution, the temperature is adjusted to 35° C., and agitation is allowed for at least 12 hours to permit the Boc deprotection reaction to occur. The reaction is monitored by HPLC or reverse phase TLC. When complete, agitation is discontinued and the phases are allowed to separate. The lower, organic phase is removed and set aside. The reactor is then charged with 12.7 Kg (9.6 L) of dichloromethane as an extraction solvent, the mixture is agitated, phases are allowed to separate, and the organic dichloromethane phase is removed. The organic extracts are combined and discarded. The remaining aqueous phase is diluted with 18.3 Kg distilled water and the temperature is raised to approximately 50° C. Distillation under vacuum (100-400 torr) is performed to remove residual dichloromethane from the reaction. The pH of the reaction is then adjusted to between 7.8-8.1 using about 9.42 Kg of 3.0N NaOH (aq) solution while keeping the temperature of the reaction below 65° C. The reaction is cooled to 50° C. and the precipitated solids are aged for at least an hour prior to cooling the mixture to room temperature. The solids are isolated by suction filtration and washed twice with 5.2 Kg portions of distilled water. The solids are dried for at least 12 hours with suction and then for an additional 12 hours in a convection oven at 55° C. The yield achieved for intermediate (23) in this example is 3.2 Kg (79%). A reactor is charged with 3.2 Kg solid intermediate (23) and the solids are suspended in 25.6 Kg of 95% ethanol as solvent. To the reactor is then added 1.1 Kg of solid D,L-malic acid (24), and the mixture is heated to reflux temperature (˜80° C.). Distilled water (˜5.7 L) is added to the reaction until a complete solution is achieved and 0.2 Kg of activated charcoal is added. The reaction mixture is passed through a filter to achieve clarification, cooled to 45° C. and held for a period of at least 2 hours to allow crystallization to occur. The reaction mixture is further cooled to 5° C. and the suspended solids are isolated by suction filtration. The solids are then washed with 6.6 KG of 95% ethanol and dried for at least 4 hours with suction under vacuum. The solids are then further dried in a convection oven for at least 12 hours at 45° C. to afford 3.1 Kg of intermediate (24) (70%). NMR (D2O, 300 MHz) δ (ppm): 8.54 (s, 1H), 7.37 (d, J=9.0 Hz, 1H), 7.05 (d, J=9.0 Hz, 1H), 4.23-4.18 (m, 1H), 4.10-3.89 (m, 1H), 3.66 (br s, 1H), 3.58 (s, 3H), 3.45 (d, J=9.0 Hz, 1H), 3.34 (d, J=9.3 Hz, 1H), 3.16 (d, J=12.9 Hz, 1H), 2.65 (dd, J=16.1, 4.1 Hz, 1H), 2.64-2.53 (m, 1H), 2.46 (dd, J=16.1, 8.0 Hz, 1H), 2.06 (br s, 1H), 1.87 (d, J=14.4 Hz, 1H), 1.58-1.45 (m, 1H), 1.15-0.95 (m, 2H), 0.91 (d, J=6.3 Hz, 3H); 0.85-0.78 (m, 2H). TLC (Whatman MKC18F Silica, 60 Å, 200 μm), Mobile Phase: 1:1 (v/v) CH3CN:0.5N NaCl (aq), UV (254/366 nm) visualization. HPLC: Mobile Phase H2O with 0.1% formic acid/Acetonitrile with 0.1% formic acid, gradient elution with 88% H2O/formic acid to 20% H2O/formic acid, Zorbax SB-C8 4.6 mm×150 mm column, Part No. 883975.906, 1.5 ml/min rate, 20 min run time, 292 nm, Detector Model G1314A, S/N JP72003849, Quat Pump Model G1311A, S/N US72102299, Auto Sampler Model G1313A, S/N DE14918139, Degasser Model G1322A, S/N JP73007229; approximate retention time for intermediate (19): 13.0 min; approximate retention time for intermediate (20): 11.6 min; approximate retention time for intermediate (21): 16.3 min; approximate retention time for intermediate (22): 18.2 min; approximate retention time for intermediate (23): 8.6 min; approximate retention time for compound (25): 8.6 min.
………………..
REF
A. ARJONA ET AL: “Nemonoxacin“, DRUGS OF THE FUTURE, vol. 34, no. 3, 1 January 2009 (2009-01-01), page 196, XP55014485, ISSN: 0377-8282, DOI: 10.1358/dof.2009.034.03.1350294
| 2 | * | ANONYMOUS: “TaiGen Announces Positive Data From the Phase II Study of Nemonoxacin (TG-873870) in Community-Acquired Pneumonia“, INTERNET CITATION, [Online] 7 April 2008 (2008-04-07), page 1, XP007919900, Retrieved from the Internet: URL:http://www.taigenbiotech.com/news.html#16> [retrieved on 2011-12-12] |
| 3 | * | ANONYMOUS: “TaiGen Biotechnology Initiates Phase II Trial Of Nemonoxacin For Treatment Of Adult Community Acquired Pneumonia (CAP)“, 20070108, [Online] 8 January 2007 (2007-01-08), page 1, XP007919910, Retrieved from the Internet: URL:http://www.taigenbiotech.com/news.html#11> [retrieved on 2011-12-12] |
| 4 | * | ANONYMOUS: “TaiGen Initiates Phase 1B Trial of a Novel Quinolone Antibiotic“, 20050618, 18 June 2005 (2005-06-18), pages 1-2, XP007919904, |
| 5 | * | See also references of WO2010002415A1 |
| WO2007110834A2 * | Mar 26, 2007 | Oct 4, 2007 | Procter & Gamble | Malate salts, and polymorphs of (3s,5s)-7-[3-amino-5-methyl-piperidinyl]-1-cyclopropyl-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid |
| WO2009023473A2 * | Aug 5, 2008 | Feb 19, 2009 | Chi-Hsin Richard King | Antimicrobial parenteral formulation |
| WO2010009014A2 * | Jul 10, 2009 | Jan 21, 2010 | Taigen Biotechnology Co., Ltd. |
|
7-4-2012
|
TREATMENT OF ANTIBIOTIC-RESISTANT BACTERIA INFECTION
|
|
|
4-18-2012
|
Coupling Process For Preparing Quinolone Intermediates
|
|
|
10-19-2011
|
Malate salts, and polymorphs of (3S,5S)-7-[3-amino-5-methyl-piperidinyl]-1-cyclopropyl-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid
|
|
|
6-18-2010
|
STEREOSELECTIVE SYNTHESIS OF PIPERIDINE DERIVATIVES
|
|
|
2-19-2010
|
PNEUMONIA TREATMENT
|
|
|
5-6-2009
|
Hydride reduction process for preparing quinolone intermediates
|
|
|
2-13-2009
|
ANTIMICROBIAL PARENTERAL FORMULATION
|
|
|
11-26-2008
|
Coupling process for preparing quinolone intermediates
|
| US8158798 | Oct 27, 2008 | Apr 17, 2012 | Taigen Biotechnology Co., Ltd. | Coupling process for preparing quinolone intermediates |
| US8211909 | Sep 8, 2008 | Jul 3, 2012 | Taigen Biotechnology Co., Ltd. | Treatment of antibiotic-resistant bacteria infection |
| WO2010002965A2 * | Jul 1, 2009 | Jan 7, 2010 | Taigen Biotechnology Co., Ltd. | Pneumonia treatmen |
WO 2007110834
WO 2007110835
WO 2007110836
WO 1999014214
WO 2010077798

1, nemonoxacin; 2, delafloxacin; 3, finafloxacin; 4, zabofloxacin; 5, JNJ-Q2; 6, DS-8587; 7, KPI-10; 8, ozenoxacin; 9, chinfloxacin; 10, ACH-702.

Onion extract slows colon cancer growth just as effectively as chemo drug

(NaturalNews) Researchers have just discovered that flavonoids extracted from common onions slow the rate of colon cancer growth in mice just as effectively as a chemotherapy drug. And while the mice on chemo saw their LDL cholesterol go up (a possible side effect of the drug), the mice on onion extract actually saw their LDL levels drop.
Onion flavonoids slow colon tumor growth by 67% in vivo
Learn more: http://www.naturalnews.com/044318_onion_extract_colon_cancer_chemotherapy_drug.html##ixzz2wD3udzfF
http://www.naturalnews.com/044318_onion_extract_colon_cancer_chemotherapy_drug.html#

FDA Asked To Improve Approval Processes For Orphan Drugs
DRUG REGULATORY AFFAIRS INTERNATIONAL
Lawmakers appealed to FDA Commissioner Margaret Hamburg to improve consistency of approval processes for orphan drugs, in a letter spearheaded by Senator Edward Markey.
“We write in recognition of the Food and Drug Administration (FDA’s) efforts to ensure public access to safe, innovative and novel therapeutics, particularly for rare diseases and where there are unmet medical needs, and to ask that you continue to commit to ensuring that potential new medicines are guided and reviewed consistently across the agency,” the letter stated, which was signed by 38 members of Congress. The lawmakers said that many families continue to struggle with limited options for rare diseases and development of new, more effective medical treatments often comes too slow. “Innovation of new and safe drugs is especially urgent for rare diseases, for which either no approved therapeutics or no cures currently exist,” the letter stated.
read
EMA publishes New Process Validation Guideline
![]()
EMA publishes New Process Validation Guideline
After the publication of the Annex 15 draft at the beginning of February 2014, the EMA made a move towards the revision of its process validation guideline. The final document was published on 27 February 2014. For a long time now, the EMA had already announced this revision in a concept paper. What’s new? click here
After the publication of the Annex 15 draft at the beginning of February 2014, the EMA made a move towards the revision of its process validation guideline. The final document was published on 27 February 2014. For a long time now, the EMA had already announced this revision in a concept paper. The objective of the revision was to integrate modern GMP aspects:
- Integration of the ICH Q8, Q9 and Q10 Guidelines
- Incorporation of Process Analytical Technology (PAT), Quality by Design (QbD) and Real-Time Release Testing (RTRT).
- Extension with regard to an “enhanced approach” and integration of “continuous process verification”
- Integration of the Annexes to the current Note for Guidance
- Harmonisation with the current FDA Guidance on Process Validation
The deadline for comments on the draft for the revision of the process validation guideline ended in October 2012 already. Now, elements in accordance with the Annex 15 have also flowed into the final document. In the following, you will read a short evaluation of the document with regard to the original draft from March 2012, the (still) applicable Note for Guidance on Process Validation and FDA’s Guidance on Process Validation. The GMP relevant aspects of the documents will also be addressed.
The original 7-page long Note for Guidance on Process Validation has more than doubled and now contains 15 pages. Even the original revision draft had only 11 pages. The change in the title to “Guideline on process validation for finished products- information and data to be provided in regulatory submissions” is noticeable. The title itself gives indication about the content of the document, namely marketing authorisation matters.
Like in the draft, the document is composed of 8 numerated chapters, a summary, definitions, references, an Annex I (Process validation scheme) and an Annex II (Standard/non-standard processes) which is a new part compared to the draft. A sub section on “Design space verification” has been newly added to the chapter on process validation.
There haven’t been big changes to the draft document released in 2012. Only the chapter “Design space verification” is brand new, all other parts have been mostly updated. The chapter on ongoing process validation has been removed. Compared to the draft, indications about standard/ non-standard processes are now available in the Annex II – like in the currently applicable Note for Guidance.
What are the changes to the currently applicable Note for Guidance on Process Validation?
Compared to the current Note for Guidance, the revision remains in its final version pretty difficult to read and rather general. This is a marketing authorisation document, which is clearly addressed in the title and only applies to finished dosage forms of chemical medicinal products for human and veterinary use but not for old ones, which are already authorised and on the market. The introduction of a validation life cycle and the integration of continued process verification (CPV) are completely new although this approach is already acquainted from ICH Q8. The “traditional approach” remains accepted. Like in the Annex 15 draft the hybrid approach remains here in the final document “nebulous”. The idea to integrate modern elements from ICH Q8, Q10 (and Q11) into the document is clearly noticeable. Yet, far less concrete references are made to ICH Q9.
A stronger overlap of the FDA Guidance would have been desirable. FDA’s Guidance also deals with APIs and biologicals, and the process validation life cycle runs like a thread through the whole FDA document. FDA’s Guidance also contains GMP aspects. The FDA Guidance explicitly addresses old products which should be integrated to stage 3 of the life cycle. Yet, there is another big difference. The revised document doesn’t highlight statistical methods like the FDA Guidance.
Before the finalisation, a comparison with the Annex 15 has been made which is a nice thing. This explains the long period between the publication of the draft (March 2012) and that of the finalisation (February 2014).
What is significant for the GMP world? On the one hand almost nothing, on the other hand quite a lot: one may wonder why? Direct references to the Annex 15 can be found with regard to the “ongoing process verification” and “concurrent validation”, which is almost nothing looking at the whole document. Moreover, validation in general is required to be executed according to the GMP regarding “continuous process verification” and “change control”; these are the essential parts of the document, and (almost) the complete document should therefore be seen from a GMP perspective.
The new EMA guideline on process validation will apply by the end of August 2014.

MEPOLIZUMAB….GSK to file severe asthma drug by year end
The first non-inhaled treatment for a difficult-to-treat form of severe asthma is getting closer to market after GlaxoSmithKline said it would initiate global filings for the drug at the end of this year, on the back of strong late-stage clinical data.
Mepolizumab – a monoclonal antibody that inhibits interleukin 5 – is being investigated as a treatment for severe eosinophilic asthma in patients who experience exacerbations despite high-dose oral or inhaled corticosteroids (ICS) and an additional controller such as long-acting beta-2 agonist.
Read more at: http://www.pharmatimes.com/Article/14-03-13/GSK_to_file_severe_asthma_drug_by_year_end.aspx#ixzz2vuANtYaK
Follow us: @PharmaTimes on Twitter
Mepolizumab (proposed trade name Bosatria) is a humanized monoclonal antibody that recognizes interleukin-5 (IL-5), and is used to treat certain kinds of asthma and white blood cell diseases.
 IL 5
IL 5
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Humanized (from mouse) |
| Target | IL-5 |
Recent studies have concluded that mepolizumab may improve exacerbations in patients with severe eosinophilic asthma, an adult-onset asthma which represents less than 5% of all asthma.
IL-5 is a chemical messenger in the immune system that stimulates the growth of eosinophils. In eosinophilic asthma, eosinophils are present in the lungs. When mepolizumab was given to people with eosinophilic asthma, it eliminated eosinophils from the bloodstream,and reduced eosinophils in the lungs and bone marrow. Mepolizumab also reduced the number of asthma exacerbations, and reduced the need for corticosteroids.[1]Mepolizumab improved the quality of life, but the improvement was “not clinically meaningful,” according to a reviewer.[2] [3]
In a recent multi-centre, double-blinded, randomised, controlled trial study of Mepolizumab in severe eosinophilic asthma, Mepolizumab reduced the number of clinically significant exacerbations compared to a placebo. Additionally Mepolizumab reduced sputum and blood eosinophil counts and was shown to be safe for up to 12 months.[4]

Mepolizumab is also in development for the management of hypereosinophilic syndrome by GlaxoSmithKline (GSK) and has received orphan drug designation by the FDA.[5] Mepolizumab has been shown to reduce the need for corticosteroids and improve symptoms in FIP1L1/PDGFRA negative hypereosinophilic syndrome.[6]
UK pharma giant GlaxoSmithKline (LSE: GSK) says that a pivotal Phase III study of mepolizumab, an investigational IL-5 antagonist monoclonal antibody, met its primary endpoint of reduction in the frequency of exacerbations, in patients with severe eosinophilic asthma.
Mepolizumab could add £400 million ($668 million) to GSK’s revenue by 2021, according to estimates from Barclays reported by The Wall Street Journal. Analysts from Deutsche Bank forecast £300 million in mepolizumab sales by 2018 for the company, already a leader in the asthma treatment sector.

The study (MEA115588) evaluated the efficacy of two-dose regimens of mepolizumab in the treatment of patients with severe eosinophilic asthma. Patients remained on their current asthma maintenance therapy throughout the study and were randomized to receive either mepolizumab 75mg intravenous (IV), 100mg subcutaneous (SC), or placebo every four weeks.
For the primary end point, both mepolizumab treatment arms showed statistically significant reductions in the frequency of clinically significant exacerbations of asthma compared to placebo (75mg IV, 47%, p<0.001; 100mg SC, 53%, p<0.001).
Adverse events reported in the study were similar across all treatment groups. The most common reported adverse events across all treatment groups were nasopharyngitis, headache, upper respiratory tract infection and asthma. The frequency of adverse events was 83% in the placebo group, 84% in the mepolizumab 75mg IV and 78% in the mepolizumab 100mg SC group. The frequency of serious adverse events was 14% in the placebo group, 7% in the mepolizumab 75mg IV and 8% in the mepolizumab 100mg SC group.
Backs up earlier studies; regulatory filing mooted at year end
Dave Allen, head of GSK Respiratory Therapy Area Unit, R&D, said: “We are really pleased to have generated further positive data on mepolizumab, consistent with the findings from our earlier exacerbation study. We now have two studies showing a reduction in exacerbations in a specific group of patients with a severe form of asthma who continue to exacerbate despite treatment with high doses of their current maintenance therapies. This is very positive news for patients. For GSK it is exciting that this is the first non-inhaled treatment for severe asthma and we will be progressing towards global filings at the end of the year.”
In addition, a second Phase III study (MEA115575) designed to evaluate the use of mepolizumab 100mg SC, every four weeks in comparison to placebo in reducing daily oral corticosteroid use while maintaining asthma control also met its primary endpoint. The study showed that patients on mepolizumab 100mg SC were able to achieve greater reductions in their maintenance oral corticosteroid dose during weeks 20-24 compared to patients on placebo (p =0.008), while maintaining asthma control.
In this study adverse events were similar across treatment groups. The most common reported adverse events in the two treatment groups were headache, nasopharyngitis, bronchitis, sinusitis, fatigue and asthma. The frequency of adverse events was 92% in the placebo and 84% in the mepolizumab treatment group. Frequency of serious adverse events was 18% in the placebo group and 1% in the mepolizumab group.
Mepolizumab Useful in Refractory Eosinophilic Asthma, a Rare Subtype of Asthma
Eosinophil.
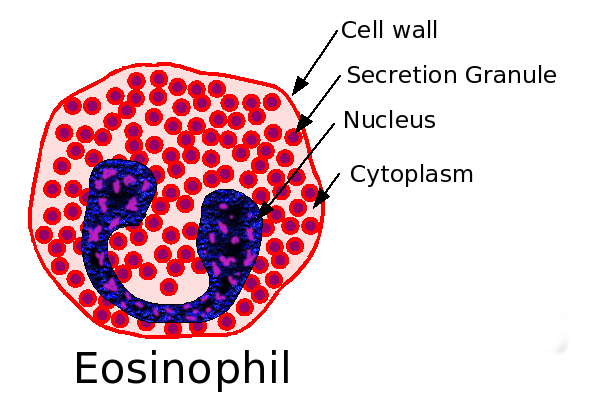
Eosinophil.Eeosinophilic form of asthma represents less than 5% of cases of adult-onset asthma and is difficult to treat.
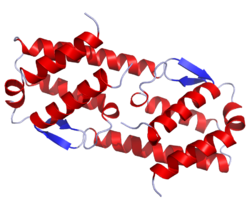
Crystal structure of human IL-5. .
Mepolizumab reduced the number of blood and sputum eosinophils and allowed prednisone sparing in patients who had asthma with sputum eosinophilia despite prednisone treatment.
Mepolizumab therapy reduced exacerbations by 43% and improved Asthma Quality of Life Questionnaire (AQLQ) scores in patients with refractory eosinophilic asthma.
Eosinophils may have a role as important effector cells in the pathogenesis of severe exacerbations of asthma in patients with eosinophilic asthma.
Cytokine targets for immunomodulators for allergic disorders.
Mediators from Eosinophils
References
- Haldar P, Brightling CE, Hargadon B, et al. Mepolizumab and exacerbations of refractory eosinophilic asthma. N Engl J Med. 2009 Mar 5;360(10):973-84.
- Nair P, Pizzichini MM, Kjarsgaard M, et al. Mepolizumab for prednisone-dependent asthma with sputum eosinophilia. N Engl J Med. 2009 Mar 5;360(10):985-93.
- Eosinophils in asthma – closing the loop or opening the door? Sally E. Wenzel, N Engl J Med. 2009 Mar 5;360(10):1026-7.
- Pavord, Ian D; Korn, Stephanie; Howarth, Peter; Bleecker, Eugene R; Buhl, Roland; Keene, Oliver N; Ortega, Hector; Chanez, Pascal (August 2012). “Mepolizumab for severe eosinophilic asthma (DREAM): a multicentre, double-blind, placebo-controlled trial”. The Lancet 380 (9842): 651–659. doi:10.1016/S0140-6736(12)60988-X.
- Phase III study of Bosatria (mepolizumab) showed disease control with reduced corticosteroid use in hypereosinophilic syndrome
- http://content.nejm.org/cgi/content/abstract/358/12/1215 Rothenberg et al 2008

FDA Approves First Device to Prevent Migraines

TUESDAY March 11, 2014, 2014 — The U.S. Food and Drug Administration on Tuesday approved the first device aimed at preventing migraines.
The device, called Cefaly, is a headband-like device that runs on a battery and sits across the forehead and over the ears, the FDA said in a statement.
“The user positions the device in the center of the forehead, just above the eyes, using a self-adhesive electrode,” the agency explained. “The device applies an electric current to the skin and underlying body tissues to stimulate branches of the trigeminal nerve, which has been associated with migraine headaches.”
Cefaly is made by Belgium-based Cefaly Technology and is available by prescription only. The device is only indicated for use by adults and should only be used for 20 minutes per day, the FDA said. The agency also noted that “the user may feel a tingling or massaging sensation where the electrode is applied.”

CEFALY DRUG-FREE MIGRAINE PAIN RELIEF
Cefaly offers patients suffering from migraine pain and headaches an efficient electrotherapeutical system delivered via an extremely comfortable, ergonomic and simple-to-use medical device.

Ann-Teresa Cusenza…..Managing Editor, Orphan Druganaut Blog

Ann-Teresa Cusenza
It is a great pleasure to write about ANN…. I read her blog everyday……………………….
Medical Information Specialist | Medical Librarian | Managing Editor, Orphan Druganaut Blog
| Current | |
|---|---|
| Previous | |
| Education |
- Ann-Teresa Cusenza, MLS, MBA
She is Managing Editor, Orphan Druganaut Blog
- read at
http://orphandruganaut.wordpress.com/this is all about orphan drugs, great work ANN
![]()

-
ABOUT | Orphan Druganaut Blog
orphandruganaut.wordpress.com/about/Ann-Teresa Cusenza, MLS, MBA. Managing Editor, Orphan Druganaut Blog. Medical Information Specialist/Pharmaceutical Competitive Intelligence Consultant.
- SPECIALITIES :
- • Providing medical library information services :
- 1. Creation of Scientific Publication Plans across therapy areas
- 2. Performed searching of medical/pharmaceutical & business databases
- 3. Performed document delivery services
- 4. Scientific literature searching and analysis
- 5. Medical fact checking
- 6. Responsible for completing research requests, adhoc requests, and large projects via phone inquiries, E-Mail, and face-to-face meetings
- • Consulting services through full Information Life Cycle :
- 1. Client consultation
- 2. Search strategy
- 3. Research
4. Information analysis and organization5. Presentation to clients
- • Literature searches and analysis using pharmaceutical/medical/healthcare and business databases, search engines, and other electronic and print resources
- • Monitoring on a daily basis, competitor products in the Drug Development Pipeline
- • Providing competitive intelligence, case scenarios, and strategic recommendations on Product Lifecycle Management in the pharmaceutical industry
- • Creating Daily Newsletters with timely information, analytic overview of pharmaceutical marketplace, analysis of medical meeting abstracts and presentations across therapy areas
- • Providing research, analysis, and identification of Domestic and International Key Opinion Leaders (KOLs) across therapy areas
- • Creation, research, writing and editing pharmaceutical/medical/healthcare Blogs using WordPress.
- COMMITMENT TO LIFELONG CONTINUING EDUCATION :
- • Emerging Web Technologies & Social Media
- • Blogging Using WordPress.
- FELLOWSHIPS :
- • National Library of Medicine (NLM) Fellowship for BioMedical Informatics at the Marine Biological Laboratory (MBL), Woods Hole, MA.
- YOU CAN CONNECT WITH HER ON TWITTER AND LINKEDIN
| Websites |
|---|
-

- Medical Librarian at HackensackUMC … ·
- Pharmaceuticals
View Ann-Teresa Cusenza’s professional profile on LinkedIn. LinkedIn is the world’s largest business network, helping professionals like Ann-Teresa Cusenza …
Check out her linkedin group

http://www.linkedin.com/groups/AnnTeresa-Cusenza-2013-Orphan-Drug-2179312.S.223691622
Thankyou Ann-Teresa Cusenza


{kind=link}