
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Scientists develop potential new drug treatment to tackle viruses

An international team of scientists have successfully developed a novel compound which early signs suggest might prevent a range of viruses from infecting humans. Researchers from Oxford, Beijing, Leeds and Innsbruck collaborated on the new inhibitor. It targets a group of viruses responsible for hand, foot and mouth disease, especially the EV71 virus. This viral group causes numerous epidemics in children, mainly in Asia, with roughly 10 million cases reported every year in China alone. Symptoms are usually mild but in some cases the disease can prove fatal – the Chinese government reported over 900 deaths in 2010. The disease is currently untreatable and is a major global threat to public health.
This discovery, published in Nature Structural and Molecular Biology, may also have important implications for combating other diseases. Hand, foot and mouth disease is caused by several closely related viruses, and the new compound is effective…
View original post 510 more words
Temozolomide 替莫唑胺

Temozolomide 替莫唑胺
Temozolomide is a DNA damage inducer.
4-methyl-5-oxo-2,3,4,6,8-pentazabicyclo[4.3.0]nona-2,7,9-triene-9-carboxamide
3,4-dihydro-3-methyl-4-oxoimidazo(5,1-d)-1,2,3,5-tetrazine-8-carboxamide
Methazolastone, Temodar, Temodal
CAS NO 85622-93-1
Molecular Weight: 194.15
MF C6H6N6O2
Cancer Research UK (Originator), Schering-Plough (Licensee), National Cancer Institute (Codevelopment)
NMR..http://file.selleckchem.com/downloads/nmr/S123702-Methazolastone-NMR-Selleck.pdf
HPLC.http://file.selleckchem.com/downloads/hplc/S123702-Methazolastone-HPLC-Selleck.pdf
Temozolomide is an antitumor agent indicated for treating patients with malignant glioma such as cancer, breast cancer, refractory anaplastic astrocytoma, i.e., patients at first relapse who have experienced disease progression in malignant glioma, glioblastoma multiform and anaplastic astrocytoma, on a drug regimen containing a nitrosourea and procarbazine.
Temozolomide preparations are sold on the US market as hard capsules containing 5 mg, 20 mg, 100 mg or 250 mg Temozolomide (marketed as Temodar® by Schering Corporation, Kenilworth, N.J., USA). In other markets it is sold as Temodal®.
Temozolomide (brand names Temodar and Temodal and Temcad) is an oral chemotherapy drug. It is an alkylating agent used for the treatment of Grade IV astrocytoma — an aggressive brain tumor, also known as glioblastoma multiforme — as well as for treating melanoma, a form of skin cancer.
Temozolomide is also indicated for relapsed Grade III anaplastic astrocytoma and not indicated for, but as of 2011 used to treatoligodendroglioma brain tumors in some countries, replacing the older (and less well tolerated) PCV (Procarbazine–Lomustine–Vincristine) regimen.
Temozolomide, 3-methyl-8-aminocarbonyl-imidazo[5,1-d]-1,2,3,5-tetrazin-4(3H)-one, is a known antitumor drug; see for example Stevens et al., J. Med. Chem. 1984, 27, 196-201, and Wang et al., J. Chem. Soc., Chem. Commun.,1994,1687-1688. Temozolomide, the compound of formula 1:

is described in U.S. Pat. No. 5,260,291 (Lunt et al.).
The synthesis of 1 by the process described in J. Med. Chem. 1984, 27, 196-201 is depicted in the scheme I below.

In this process, 5-amino-1H-imidazole-4-carboxamide (A) is converted into 5-diazo-1H-imidazole-4-carboxamide (B), which is then cyclized with methylisocyanate in dichloromethane to provide a high yield of temozolomide. However, this process requires isolation of the unstable and potentially dangerous 5-diazo-1H-imidazole-4-carboxamide (B). Moreover, methylisocyanate is a difficult reagent to handle and ship, especially on the industrial scale, and indeed is better avoided in industrial manufacture. Furthermore, the cycloaddition of methylisocyanate requires a very long reaction time: Table I in J. Med Chem.1984, 27,196-201, suggests 20 days. Additionally, Stevens et al mention that the cycloaddition of the methylisocyanate to the compound of the formula (B) can proceed through two different intermediates:
The production of I by the two processes described in J. Chem. Soc., Chem. Commun., 1994, 1687-1688 provides a low overall yield from 5-amino-1H-imidazole-4-carboxamide (A): less than 20% (unoptimized—about 17% through 5-diazo-1H-imidazole-4-carboxamide (B) and about 15% through 5-amino-N1-(ethoxycarbonylmethyl)-1H-imidazole-1,4-dicarboxamide (C)); Scheme II below

The agent was developed by Malcolm Stevens[1] and his team at Aston University in Birmingham,[2][3] Temozolomide is a prodrug and animidazotetrazine derivative of the alkylating agent dacarbazine. It has been available in the US since August 1999, and in other countries since the early 2000s.
The therapeutic benefit of temozolomide depends on its ability to alkylate/methylate DNA, which most often occurs at the N-7 or O-6 positions ofguanine residues. This methylation damages the DNA and triggers the death of tumor cells. However, some tumor cells are able to repair this type of DNA damage, and therefore diminish the therapeutic efficacy of temozolomide, by expressing a protein O6-alkylguanine DNA alkyltransferase (AGT) encoded in humans by the O-6-methylguanine-DNA methyltransferase (MGMT) gene.[4] In some tumors, epigenetic silencing of the MGMT gene prevents the synthesis of this enzyme, and as a consequence such tumors are more sensitive to killing by temozolomide.[5] Conversely, the presence of AGT protein in brain tumors predicts poor response to temozolomide and these patients receive little benefit from chemotherapy with temozolomide.[6]
- Nitrosourea- and procarbazine-refractory anaplastic astrocytoma
- Newly diagnosed glioblastoma multiforme
- Malignant prolactinoma
Temozolomide (sometimes referred to as TMZ) is an imidazotetrazine derivative of the alkylating agent dacarbazine. It undergoes rapid chemical conversion in the systemic circulation at physiological pH to the active compound, 3-methyl-(triazen-1-yl)imidazole-4-carboxamide (MTIC). Temozolomide exhibits schedule-dependent antineoplastic activity by interfering with DNA replication. Temozolomide has demonstrated activity against recurrent glioma. In a recent randomized trial, concomitant and adjuvant temozolomide chemotherapy with radiation significantly improves, from 12.1 months to 14.6 months, progression free survival and overall survival in glioblastoma multiforme patients.
Formulations
Temozolomide is available in the United States in 5 mg, 20 mg, 100 mg, 140 mg, 180 mg & 250 mg capsules. Now also available in an IV form for people who can not swallow capsules or who have insurance that does not cover oral cancer agents.
A generic version is available in the UK.
Further improvement of anticancer potency
Laboratory studies and clinical trials are investigating whether it might be possible to further increase the anticancer potency of temozolomide by combining it with other pharmacologic agents. For example, clinical trials have indicated that the addition of chloroquine might be beneficial for the treatment of glioma patients.[8] In laboratory studies, it was found that temozolomide killed brain tumor cells more efficiently when epigallocatechin gallate (EGCG), a component of green tea, was added; however, the efficacy of this effect has not yet been confirmed in brain tumor patients.[9]More recently, use of the novel oxygen diffusion-enhancing compound trans sodium crocetinate (TSC) when combined with temozolomide and radiation therapy has been investigated in preclinical studies [10] and a clinical trial is currently underway.[11]
Because tumor cells that express the MGMT gene are more resistant to killing by temozolomide, it was investigated[according to whom?] whether the inclusion of [[O6-benzylguanine]] (O6-BG), an AGT inhibitor, would be able to overcome this resistance and improve the drug’s therapeutic effectiveness. In the laboratory, this combination indeed showed increased temozolomide activity in tumor cell culture in vitro and in animal models in vivo.[12] However, a recently completed phase-II clinical trial with brain tumor patients yielded mixed outcomes; while there was some improved therapeutic activity when O6-BG and temozolomide were given to patients with temozolomide-resistant anaplastic glioma, there seemed to be no significant restoration of temozolomide sensitivity in patients with temozolomide-resistant glioblastoma multiforme.[13]
There are also efforts to engineer hematopoietic stem cells expressing the MGMT gene prior to transplanting them into brain tumor patients. This would allow for the patients to receive stronger doses of temozolomide, since the patient’s hematopoietic cells would be resistant to the drug.[14]
High doses of temozolomide in high grade gliomas have low toxicity, but the results are comparable to the standard doses.[15]
A case report suggests that temozolomide may be of use in relapsed primary CNS lymphoma.[16] Confirmation of this possible use seems indicated.
Temozolomide, 3-methyl-8-aminocarbonyl-imidazo[5,1-d]- 1 ,2,3,5-tetrazin- 4(3H)-one, is a known antitumor drug; see for example Stevens et al., J. Med. Chem. 1984, 27, 196-201 , and Wang et al., J. Chem. Soc, Chem. Commυn., 1994, 1687-1688. Temozolomide, the compound of formula 1 :

1 is described in U.S. Patent No. 5,260,291 (Lunt et al.).
The synthesis of 1 by the process described in J. Med. Chem. 1984, 27, 196- 201 is depicted in the scheme I below. Scheme I:

In this process, 5-amino-1 H-imidazole-4-carboxamide (A) is converted into 5- diazo-1 H-imidazole-4-carboxamide (B), which is then cyclized with methylisocyanate in dichloromethane to provide a high yield of temozolomide.
However, this process requires isolation of the unstable and potentially dangerous 5-diazo-1 H-imidazole-4-carboxamide (B). Moreover, methylisocyanate is a difficult reagent to handle and ship, especially on the industrial scale, and indeed is better avoided in industrial manufacture.
Furthermore, the cycloaddition of methylisocyanate requires a very long reaction time: Table I in J. Med Chem. 1984, 27,196-201 , suggests 20 days. Additionally, Stevens et al mention that the cycloaddition of the methylisocyanate to the compound of the formula (B) can proceed through two different intermediates:
The production of I by the two processes described in J. Chem. Soc, Chem.
Commun., 1994, 1687-1688 provides a low overall yield from 5-amino-1 H- imidazole-4-carboxamide (A): less than 20% (unoptimized – about 17% through 5- diazo-1 H-imidazole-4-carboxamide (B) and about 15% through 5-amino-N1– (ethoxycarbonylmethyl)- 1 H-imidazole- 1 ,4-dicarboxamide (C)); Scheme II below
Scheme II:

Moreover, the unstable 5-diazo-1 H-imidazole-4-carboxamide (B) still has to be isolated in the branch of this process that uses it as an intermediate. Clearly, therefore, there is a need for synthetic methods that: a) are more convenient and higher yielding, especially on commercial scale; b) approach the synthesis of the temozolomide nucleus in novel ways; or c) improve the preparation or use of intermediates for the processes.
Temozolomide of formula I, is an antitumor drag and is chemically known as 3-methyl-8- aminocarbonyl-imidazole[5,l-d]-l,2,3,5-tetrazin-4(3H)-one.

Formula I
It is indicated for treating patients with malignant glioma such as cancer, breast cancer, refractory anaplastic, astrocytoma, i.e. patient at first relapse who have experienced disease progression in malignant glioma, glioblastoma multiform and anaplastic astrocytoma, on a drug containing a nitrosourea and procarbazine. It is sold in the US market as hard capsules containing 5 mg, 20 mg, 100 mg or 250 mg as Temodar® by Schering corporation.
Temozolomide and compounds having similar activity (higher alkyl analogues at the 3 -position) were first disclosed in US patent 5,260,291. According to said patent, temozolomide is prepared by the reaction of 5-diazoimidazole-4-carboxamide with methyl isocyanate in the presence of N- methylpyrrolid-2-one in dichloromethane at room temperature for three to four weeks. Melting point of temozolomide reported in above patent is 200 0C (recrystallized from acetonitrile); 21O0C with effervescence (recrystallized from acetone and water), and 2150C with effervescence and darkening (recrystallized from hot water). Major drawback of process is the longer reaction duration of three to four weeks for completion of reaction.
Further, the process described in the patent involves use of low boiling and extremely toxic, methyl isocyanate, which is very difficult to handle, especially on industrial scale, as its use should be avoided in the industrial synthesis. Further, cycloaddition reaction requires a very long period of 21 to 28 days, which makes the process unattractive for industrial scale.
US patent 5,003,099 discloses a process for preparation of aminocyanoacetamide, a key intermediate for the synthesis of temozolomide. According to the patent, aminocyanoacetamide is synthesized in two steps by the reaction of cyanoacetic acid alkyl ester using sodium nitrite in the presence of glacial acetic acid to form a hydroxyimino intermediate, which is then reduced in the presence of platinum on carbon to yield aminocyanoacetic acid alkyl ester, which is unstable.
The alkyl ester intermediate is then in situ reacted with aqueous ammonia to give the desired product. The main drawback of the above mentioned process is the use of aqueous ammonia, since aminocyanoacetamide, generated in reaction, is soluble in aqueous solution and hence difficult to extract from the reaction mass which results in lower yields. The patent is silent about the purity of intermediate and process needs extraction of the above mentioned intermediate from filtrate.
US patent 6,844,434 describes synthesis of temozolomide by cyclization of 5-amino-l-(N-rnethyl- hydrazinocarbonyl)-lH-imidazole-4-carboxylic acid in the presence of tetrabutyl nickel and periodic acid to form a reaction mixture which is concentrated under reduce pressure and resulting residue was treated with acetonitrile and filtered. The filtrate was concentrated and chromatographed on a column of silica gel to give temozolomide.
Use of time consuming and cumbersome technique i.e. column chromatography for isolation of product makes the process not suitable to employ at industrial level. US patent 7,087,751 discloses a process for the preparation of temozolomide from protected imidazole intermediate.
The process involves reaction of l-methyl-3-carbamoyliminomethyl-urea with JV- protected aminocyanoacetamide in the presence of acetic acid in a suitable solvent to form an JV- protected imidazole intermediate which is then cyclized in the presence of lithium chloride to minimize undesired cyclisation product. After cyclisation, the protected group has to be removed which makes the process more laborious with more number of steps.
As exemplified in example 1 of the above patent, yield of the JV-protected imidazole intermediate obtained is very low, almost half of the product goes in the filtrate which further needs extraction from the filtrate. After extraction of inteπnediate from the filtrate, the combined yield is only 67 %. The intermediate obtained is only 93 to 94% pure and requires additional purifications, crystallization using ethyl acetate and slurry wash with mixture of methyl tertiary butyl ether and isopropanol. These additional purification further takes away around 20 % yield of the inteπnediate thus yield of the pure intermediate, which is suitable for the further reaction, remains around 53 % which is very low from commercial point of view.
The patent also describes condensation of l-methyl-3-carbamoyliminomethyl-urea with unprotected aminocyanoacetamide in presence of acetic acid to give an imidazole intermediate. This patent fails to disclose the process of conversion of above imidazole intermediate to temozolomide, but only up to hydrolysis to prepare 5-amino-lH-imidazole-4-carboxamide hydrochloride is reported.
Another US patent no. 6,844,434 of same applicant (Schering) discloses a process for the conversion of 5-amino- lH-imidazole-4-carboxamide hydrochloride, which is prepared by the hydrolysis of above imidazole intermediate, to temozolomide. By combining the above two processes, this adds further four additional steps to the synthesis of temozolomide. The process of preparation of temozolomide is described by the following scheme:

It has been observed that for the preparation of unprotected imidazole intermediate as exemplified in US 7,087,751, use of excess amount of the acetic acid (around 21 times with respect to aminocyanoacetamide) is reported. Thereafter acetic acid is removed by distillation.
The inventors of the present invention have repeated example 2 as described in US 7,087,751 for the preparation of unprotected imidazole intermediate. As per the process, after the completion of the reaction, acetic acid has to be removed from the reaction mixture. It is noticed that removal of acetic acid is a very tedious move so as on commercial scale and leads to decomposition.
In a publication namely, Journal of Organic Chemistry, volume 62, no. 21, 7288-7294, a process is disclosed for the preparation of temozolomide by the hydrolysis of 8-cyano-3-methyl-[3H]-imidazole~ [5,l-d]-tetrazin-4-one in the presence of hydrochloric acid to give hydrochloride salt of temozolomide, which has to be neutralized to obtain temozolomide. In the same Journal, another process for the preparation of temozolomide is also described. Temozolomide is prepared by the nitrosative cyclization of imidazole intermediate using aqueous solution of sodium nitrite and tartaric acid to give temozolomide in 45 % yield in solution.
US patent publication 2007/0225496 exemplified a process for preparation of temozolomide by pyrolising N’-methyl-N,N-diphenyl urea to form vapor of methyl isocyanate which is then reacted with 5-diazo-5H-imidazole-4-carboxylic acid amide to form temozolomide.
The above described process involves use of methyl isocyanate, which is highly flammable and makes the process unsuitable for industrial synthesis, hi addition to this, isolation of temozolomide from the reaction mixture requires addition of large amount of ethyl acetate followed by addition of hexane and again ethyl acetate to isolate compound.
US patent publication 2009/0326028 describes a process for preparation of temozolomide by diazotization of imidazole intermediate in the presence of at least one metal halide, a source of nitrous acid and an acid to form acidic solution of temozolomide, wherein temozolomide forms a salt with acid. The desired product i.e. temozolomide is then isolated from the acidic solution by extraction with a solvent.
The process requires very strict reaction parameters including the addition of metal halide during diazotization as well as addition of pre-cooled reaction mixture to sodium nitrite solution to achieve desired level of selective cyclization. Patent application also describes two methods for the extraction of temozolomide.
US patent publication 2010/0036121 discloses a process for the preparation of temozolomide by reaction of 5-aminoimidazole-4-carboxamide with N-succinimidyl-N’-methylcarbamate to form carbamoyl 5~aminoimidazole-4-carboxamide which is then reacted with alkali or alkaline earth nitrile to give reaction mass containing temozolomide
-
Temozolomide, is a known antitumour drug, and is represented by formula I:

3-methyl-8-aminocarbonyl-imidazo [5,1-d]-1,2,3,5-tetrazin-4(3H)-one
-
It is described in US 5,260,291 together with compounds of broadly similar activity such as higher alkyl analogs at the 3-position.
-
J.Med.Chem. 1984, 27, 196-201 describes a process wherein 5-amino-1H-imidazole-4-carboxamide is converted into 5-diazo-1H-imidazole-4-carboxamide, which is then cyclised with methylisocyanate in dichloromethane to provide a high yield of temozolomide.
-
This process requires isolation of the unstable and potentially dangerous 5-diazo-1H-imidazole-4-carboxamide, methyl isocyanate is a difficult reagent to handle and ship, especially on the industrial scale. Furthermore, the cycloaddition of methylisocyanate requires a long reaction time (Table I in J.Med.Chem. 1984, 27, 196-201, suggests 20 days).
-
The product obtained by this process contains, high residual dichloromethane. It is essential to limit dichloromethane content in the final API below 600 ppm as per ICH guideline. Dichloromethane content can be reduced if one follows technique of US 5,260,291 .
-
US 5,260,291 discloses acetone-water recrystallisation of temozolomide, which results in low yield (60% recovery) due to decomposition of temozolomide to impurities like 5-(3-methyltriazen-1-yl)imidazole-4-carboxamide, compound of formula V

and 5-amino-1H-imidazole-4-carboxamide.
-
The production of compound of formula I by the two processes described in J.Chem.Soc., Chem.Commun., 1994, 1687-1688 provides a low overall yield from 5-amino-1H-imidazole-4-carboxamide: less than 20% (about 17% through 5-diazo-1H-imidazole-4-carboxamide and about 15% through 5-amino-N1-(ethoxy carbonylmethyl)-1H-imidazole-1,4-dicarboxamide).
-
The unstable 5-diazo-1H-imidazole-4-carboxamide has to be isolated in the branch of this process that uses it as an intermediate.
-
US 2002/0133006 discloses a process for the preparation of compound of formula I using methyl hydrazine which is a toxic and flammable liquid, hence not feasible on industrial scale and the final isolation involves tedious workup including column chromatography.
-
J.Org.Chem. 1997, 62, 7288-7294 describes a process wherein the final step of diazotization provides equi-formation of aza-hypoxanthine and temozolomide, resulting in low yield. This literature does not provide the experimental procedure for work up.
-
US 2005/0131227 describes a process involving the use of a bulky protecting group on nitrogen of the primary amide for cyclisation in presence of LiCl to minimize the undesired cyclization product. After cyclization the protecting group has to be removed which makes the process more laborious with more number of steps (Scheme I).

U.S. Pat. No. 6,844,434 describes the preparation of Temozolomide, alkyl analogs and intermediates thereof. The process, which is depicted in Scheme 3 below, comprises reacting 5-amino-1H-imidazole-4-carboxamide hydrochloride (II) with 4-nitrophenyl chloroformate to afford compound (III), which is subsequently reacted with methyl hydrazine to obtain the corresponding compound (IV), which is cyclized to yield Temozolomide.

Another process for preparing Temozolomide is described in U.S. patent application having the Publication No. 2002/0095036 (see Scheme 4 below). In this process, the imine (V) is converted to 2-cyano-N-(1,1-dimethylethyl)-2-[(diphenyl-methylene)amino]-acetamide, which is converted to 2-amino-2-cyano-N-(1,1-dimethyl-ethyl)-acetamide hydrochloride.
The latter is reacted with compound (VI) to obtain 5-amino-N4-(1,1-dimethylethyl)-N1-methyl-1H-imidazole-1,4-dicarboxamide, which is converted to 3,4-dihydro-N-(1,1-dimethylethyl)-3-methyl-imidazo-[5,1-d]-1,2,3,5-tetrazine-8-carboxamide (tert-butyl-Temozolomide), which yields Temozolomide under acidic treatment with concentrated sulfuric acid.

Yet another synthesis of Temozolomide is described by Stevens et al. in J. Org. Chem., Vol. 62, No. 21, 7288-7294, 1997, wherein Temozolomide hydrochloride salt is obtained in 65% yield by the hydrolysis of 8-cyano-3-methyl-[3H]-imidazo-[5,1-d]-tetrazin-4-one with hydrochloric acid, as shown in Scheme 5.

The main disadvantage of this process is the low yield in which Temozolomide hydrochloride is obtained (65%). It is assumed that the relatively elevated temperature of 60° C. used in the process increases the content of decomposition products.






…………………………
Synthesis
US Patent 8,232,392
Temozolomide (1) is a drug that was discovered more than 30 years ago. In the past 10 years, it has been used to treat aggressive brain tumors. S. Turchetta and co-inventors summarize several processes for preparing temozolomide, all of which use toxic reagents such as MeNCO or MeNHNH2or generate large amounts of chemical waste. They describe a safer route to 1.
The inventors’ method starts with the preparation of carbamoyl compound 4 from amide 2 by treating it with succinimidyl reagent 3 in the presence of a base. The product is isolated in 88% yield and 96.9% purity by HPLC. Reagent 3 is a nonexplosive, crystalline solid with comparatively low toxicity and is much safer than MeNCO for this reaction.

In the next stage, the amine group in 4 is converted to diazonium salt 5 via a diazotization reaction. The details of this reaction are not described, but reference is made to a method reported in 1997 (Wang, Y., et al. J. Org. Chem. 1997, 62, 7288–7294). Compound 5 is not isolated; when acid is added, it cyclizes by the reaction of the diazonium group with one of the two amide groups to give products 1 and 6 in approximately equal amounts. The desired product 1 is formed by the reaction of the secondary amide group; when the primary amide reacts, the product is its isomer, 6.
Products 1 and 6 are separated by passing the acidified reaction mixture from the diazotization reaction over a column of a polymeric adsorbent resin. The material used in the example is XAD 1600 from Rohm & Haas; other resins are covered in the claims. Compound 6 elutes from the column first; then 1 is eluted with acidified aq EtOH. After separation, 1 is recrystallized from acidified acetone and isolated in 30% yield with 99.9% purity.
The process provides an alternative, safer route to temozolomide, but half of intermediate 4 is lost as unwanted product 6. [Chemi S.p.A. [Cinisello Balsamo, Italy]. US Patent 8,232,392, July 31, 2012; )
………………..
SYNTHESIS
http://www.google.com/patents/WO2002057268A1?cl=en

EXAMPLE 1
Preparation of Temozolomide (1 ) Step A Preparation compound (3)

5-Amino-1 H-imidazole-4-carboxamide*HCI (4) (25 g, 0.154 mol) (Aldrich 16,496-8), CH2CI2 (0.6 L) and Et3N (45 mL) (Aldrich, 13,206-3) were placed into a dry 2-liter, three-necked flask equipped with dropping funnel, a gas inlet tube, a gas outlet tube, reflux condenser and mechanical stirrer, and maintained under a positive pressure of nitrogen at ambient temperature. The mixture was stirred, and a solution of 400 mL of 4-nitrophenyl chloroformate (34 g, 0.169 mol) (Aldrich, 16,021-0) in CH2CI2was added dropwise.
The reaction mixture was stirred vigorously for 4 hours and then left to stand for 18 hours at room temperature. The precipitate was collected by vacuum filtration and washed with H20 (1.5 L) to afford the product (3) as a pale yellow solid (42 g, 0.144 mol). 1H NMR (400MHz, DMSO-d6, δ): 8.40 (d, 2H), 7.83 (s, 1 H), 7.74 (d, 2H), 7.08 (bs, 1 H), 6.95 (bs, 1 H), 6.52 (s, 2H). Step B Preparation of compound (2)

Compound (3) (42 g, 0.144 mol) and DMF (0.27 L) were placed into a dry
1 -liter, three-necked flask equipped with dropping funnel, a gas inlet tube, a gas outlet tube, reflux condenser and mechanical stirrer, and maintained under a positive pressure of nitrogen. The reaction mixture was cooled to 0°C, and methylhydrazine (10 mL, 0.188 mol) (Aldrich, M5.000-1 ) was added dropwise.
The reaction mixture was stirred vigorously for 1 hour at 0°C and was then poured into EtOAc (2.1 L). The precipitate was collected by vacuum filtration and was dried under vacuum (20 mm Hg, room temperature, 18 hours) to afford (2) as a tan solid (27.1 g, 0.137 mol). 1H NMR (400MHz, DMSO-d6, δ): 7.62 (s, 1 H), 6.85 (bs, 1 H), 6.75 (bs,1 H), 6.00 (s, 2H), 5.10 (s, 2H), 3.15, s, 3H).mp: 188°C (dec).
Analysis: Calcd for C6H10N6O2: C, 36.36; H, 5.09; N, 42.41.
Found: C, 36.46; H, 4.99; N, 42.12.
Step C Preparation of Temozolomide (1 )

2 1 (Temozolomide)
Compound (2) (500 mg, 2.5 mmol), Bu4NI (95 mg, 0.25 mmol), THF (250 mL) and CH3CN (250 mL) were placed into a dry 1 -liter, three-necked flask equipped with dropping funnel, a gas inlet tube, a gas outlet tube, reflux condenser and mechanical stirrer, and maintained under a positive pressure of nitrogen.
The reaction mixture was heated at 60°C for 20 mm and then cooled to room temperature. H5lθ6 (1.14 g, 5 mmol) was added and the reaction mixture was stirred vigorously at room temperature for 1 hour. The resulting solution was treated with saturated aqueous Na2S2O3 (5 mL) and was then concentrated under reduced pressure to dryness. The residue was treated with CH3CN (200 mL) and was filtered. The filtrate was concentrated and chromatographed on a column of silica gel (1.5% to 2% AcOH/EtOAc) to afford temozolomide (1 ) (280 mg). 1H NMR (400MHz, DMSO-d6, δ): 8.80 (s, 1 H), 7.80 (bs, 1 H), 7.66 (bs, 1 H), 3.43 (s,3H).
………………
SYNTHESIS

…………………
SYNTHESIS
http://www.google.com/patents/WO2010140168A1?cl=en
Accordingly, the present invention provides an improved process for the preparation of temozolomide of formula I,

Formula I which proves to be efficient and industrially advantageous.
The process comprises the step of: a), condensing compound of formula II,

Formula II with compound of formula III,
CH3 H CH3 Formula III in the presence of an acid in an alcoholic solvent to form a compound of formula IV;

Formula IV b). isolating the compound of formula IV from the reaction mixture by filtration; c). diazotizing and cyclizing the compound of formula IV in the presence of source of nitrous acid and a suitable acid; d). isolating temozolomide therefrom; and e). optionally purifying temozolomide of formula I.
Accordingly, the present invention provides an improved process for the preparation of temozolomide of formula I, process comprises the steps of: a), diazotizing and cyclizing the compound of formula IV in the presence of a source of nitrous acid and a suitable acid; b). optionally, cooling the reaction mixture; c). isolating precipitate of temozolomide from the reaction mixture; and d). purifying temozolomide of formula I with a suitable solvent
REFERENCE EXAMPLE:
Preparation* of S-Aøiino-N’-methyl-lH-imidazole-ljΦdicarboxamide (US 7,087,751) 2-Amino-2-cyanoacetamide (10 g), l-methyl-3-methylcarbamoyliminomethyl urea (19 g) and acetic acid (120 ml) were stirred together at ambient temperature under the positive pressure of nitrogen for 2 hours. Excess acetic acid was removed under reduced pressure and methyl tertiary butyl ether (25 ml) was added to the concentrated reaction mass, cooled to obtained crude solid.
The mixture was stirred for 30 minutes and the precipitate was collected by vacuum filtration. The solid was dried under vacuum at 20-250C for 18 hours to obtain 13 g of title compound as grayish solid. The crude product was stirred with water (66 ml) for 1 hour at 20-250C, filtered, suck dried and dried under vacuum at2O0C for 18 hours to obtain 11.2 g of title compound as greyish solid.
EXAMPLES
Example 1: Preparation of hydroxylirainocyano acetic acid ethyl ester
To a suspension of ethyl cyanoacetate (1.0 Kg, 8.84 mol) and sodium nitrite (0.735 kg, 10.65 mol) in water (0.80 L), acetic acid (0.70 kg, 11.66 mol) was added at 0-50C over a period of one hour.
Temperature was slowly raised to 23-270C and the reaction mixture was stirred for one hour at that temperature. After the complete consumption of ethyl cyanoacetate (monitored by TLC/GC), the reaction mixture was extracted with ethyl acetate (5 x 1.5 L). The combined organic layer was successively washed with 10% sodium bicarbonate (2 x 1.25 L) and brine solution (1.25 L), dried over sodium sulfate and filtered through hyflow bed. Solvent was removed under reduced pressure at 40-
450C. The resulting solid was stirred with cyclohexane (3.0 L) for 30 minutes at 25-300C, filtered and dried at 40-450C under vacuum to afford 1.14 kg (91.2 %) of title compound having purity 99.82% by
HPLC.
Example 2: Preparation of aminocyanoacetic acid ethyl ester
To a solution hydroxyliminocyano acetic acid ethyl ester (1.14 Kg, 8.02 mol) in methanol (11.4 L) was added 5% platinum on carbon (91.2 g, 50 % wet) and the mixture was hydrogenated at hydrogen gas pressure of 6.2-6.4 kg/cm2 over a period of 12 hours and the completion of reaction was checked by
TLC. The reaction mixture was filtered under nitrogen atmosphere to recover the catalyst. The filtrate was used as such for the next stage.
Example 3: Preparation of amimøcyanoacetamide
The solution of aminocyanoacetic acid ethyl ester (as prepared above) in methanol was cooled to 0-5
0C and ammonia gas was purged into it approximately for 1 hour. After the completion of the reaction
(monitored by TLC), the reaction mass was concentrated to 2.5-3.0 L under reduced pressure at 40-
45°C, cooled to 0-50C and stirred for 1 hour. The precipitated solid was filtered, washed with chilled methanol (200 ml) and dried at 35-400C under vacuum for 6 hours to obtain 572 g of title compound.
The resulting product was added to methanol (4.57 L) and heated to reflux till the solution become clear. Activated charcoal (25g) was added to the reaction mixture and refluxed for 15 minutes. The solution was filtered through hyflow bed, the bed was washed with methanol (500 ml) and the filtrate was concentrated to half of its original volume (approx 2.0 L). The mixture was cooled to 0-50C and stirred for 45 minutes. The resulting solid was filtered, washed with chilled methanol (250 ml) and dried at 40-450C under vacuum to obtain 425g (53.6%) of pure title compound having purity 99.46% by HPLC. Example 4: Preparation of l-methyl-3-methylcarbamoyliminomethyl urea
A suspension of monomethyl urea (1.5 kg, 20.27 mol) in triethyl orthoformate (4.5 L, 30.40 mol) was heated to reflux at 150-1600C for 12 hours. The reaction mixture was cooled to 5-100C, and stirred for 1 hour to ensure complete precipitation, of the product. The resulting solid was filtered, washed with ethyl acetate (350ml) and dried under vacuum at 45-5O0C to yield 1.08 kg (67.9%) of title compound having purity 93.82% by HPLC.
Exainple-5: Preparation of S-amino-N^methyl-lH-imidazole-l^-dicarboxamide Acetic acid (200 ml, 3.53 mol) was added to a suspension of aminocyanoacetamide (40Og, 4.04 mol) and l-methyl-3-methylcarbamoyliminomethyl urea (76Og, 4.8 mol) in methanol (2.0 L) at 20-250C and the mixture was stirred at 20-250C for 18 hours till completion of the reaction (monitored by HPLC). The reaction mixture was cooled to 0-50C, stirred for 1 hour and the resulting solid was filtered, washed with chilled methanol (450 ml), suck dried and finally dried under vacuum at 30-350C to afford 648 g (88.04%) of title compound as an off white colored solid having purity 99.21 % by HPLC. Example 6: Preparation of temozolomide
Acetic acid (450 ml, 7.95 mol) was added to a suspension of S-amino-N^methyl-lH-imidazole-l^- dicarboxamide (500g, 2.73mol) and sodium nitrite (25Og, 3.62mol) in water (5.0 L) at -5 to 00C at such a rate so that temperature does not rise above 5°C. The reaction mixture was stirred at 0 to 5°C for one hour and absence of starting material was checked by HPLC analysis. Ice bath was removed and powdered calcium chloride (1.25Kg) was added in small lots to the reaction mass and stirred at 25- 300C for 2 hours. The reaction mass was extracted with a 2.5% solution of dimethylsulfoxide in dichloromethane (5 X 50 L). Combined organic layer was dried over sodium sulfate and filtered through a hyflow bed. Solvent was removed under reduced pressure below 4O0C and residual dimethylsulfoxide layer was degassed completely. The dimethylsulfoxide layer was cooled to 0 to – 100C and stirred for 1 hour. The resulting solid was filtered, washed with ethyl acetate (25OmL), and suck dried for 2 hours to afford 32Og of the title compound having purity 78.5% by HPLC. Example 7: Preparation of temozolomide
Acetic acid (9ml, 0.159mol) was added to a suspension of 5-ammo-N1 -methyl- lH-imidazole- 1,4- dicarboxamide (1Og, 0.054mol) and sodium nitrite (5g, 0.072mol) in water (100ml) at -5 to 00C at a rate so that temperature does not rise above 0-50C. The reaction mixture was stirred at 0-50C for one and half hour. Brine (30g) was added to the reaction mixture and stirred at room temperature for two hours to saturate the reaction mixture. The reaction mass was extracted with a 2.5% solution of dimethylsulfoxide in dichloromethane (5 X 1 L). Combined organic layer was dried over sodium sulfate and filtered through a hyflow bed. Solvent was removed under reduced pressure and residual dimethylsulfoxide layer was degassed completely. The dimethylsulfoxide layer was cooled to 0 to -5°C and stirred for 1 hour. The resulting solid was filtered, washed with ethyl acetate (2x 5 ml), and suck dried for 2 hours to afford 5.0 g of the title compound having purity 81.6% by HPLC. Example 8: Preparation of temozolomide
Acetic acid (450ml) was added to a suspension of 5 -amino-N1 -methyl- lH-imidazole- 1,4- dicarboxamide (500g) and sodium nitrite (25Og) in water (5.0 L) at -5 to O0C at a rate so that temperature does not rise above 0-50C. The reaction mixture was stirred at 0-50C for one and half hour and the absence of starting material was checked by HPLC analysis. Ice bath was removed and powdered calcium chloride (1.25 kg) was added to the reaction mixture and stirred at room temperature for two hours. The reaction mass was extracted with a 2.5% solution of dimethylsulfoxide in dichloromethane (5 X 50 L). Combined organic layer was dried over sodium sulfate and filtered through a hyflo bed. Solvent was removed under reduced pressure at below 400C and residue at 35- 400C was filtered through a candle filter to remove suspended particles and the filtrate was then degassed completely. The residual dimethylsulfoxide layer was cooled to 0±2°C and stirred for one hours. The resulting solid was filtered and sucked dried. The solid was then washed with ethyl acetate (2x 250 ml), and suck dried for 1 hours to afford 240 g of the title compound.
………………………………….
SYNTHESIS
http://www.google.com/patents/US20020133006
Example 1
Preparation of Temozolomide (1)

5-Amino-1H-imidazole-4-carboxamide.HCl (4) (25 g, 0.154 mol) (Aldrich 16,496-8), CH2Cl2(0.6 L) and Et3N (45 mL) (Aldrich, 13,206-3) were placed into a dry 2-liter, three-necked flask equipped with dropping funnel, a gas inlet tube, a gas outlet tube, reflux condenser and mechanical stirrer, and maintained under a positive pressure of nitrogen at ambient temperature. The mixture was stirred, and a solution of 400 mL of 4-nitrophenyl chloroformate (34 g, 0.169 mol) (Aldrich, 16,021-0) in CH2Cl2 was added dropwise. The reaction mixture was stirred vigorously for 4 hours and then left to stand for 18 hours at room temperature. The precipitate was collected by vacuum filtration and washed with H2O (1.5 L) to afford the product (3) as a pale yellow solid (42 g, 0.144 mol).
1H NMR (400 MHz, DMSO-d6, δ): 8.40 (d, 2H), 7.83 (s, 1H), 7.74 (d, 2H), 7.08 (bs, 1H), 6.95 (bs, 1H), 6.52 (s, 2H).

Compound (3) (42 g, 0.144 mol) and DMF (0.27 L) were placed into a dry 1-liter, three-necked flask equipped with dropping funnel, a gas inlet tube, a gas outlet tube, reflux condenser and mechanical stirrer, and maintained under a positive pressure of nitrogen. The reaction mixture was cooled to 0° C., and methylhydrazine (10 mL, 0.188 mol) (Aldrich, M5,000-1) was added dropwise. The reaction mixture was stirred vigorously for 1 hour at 0° C. and was then poured into EtOAc (2.1 L). The precipitate was collected by vacuum filtration and was dried under vacuum (20 mm Hg, room temperature, 18 hours) to afford (2) as a tan solid (27.1 g, 0.137 mol).
1H NMR (400 MHz, DMSO-d6, δ): 7.62 (s, 1H), 6.85 (bs, 1H), 6.75 (bs,1H), 6.00 (s, 2H), 5.10 (s, 2H), 3.15, s, 3H).mp: 188° C. (dec.).
Analysis: Calcd for C6H10N6O2: C, 36.36; H, 5.09; N, 42.41.
Found: C, 36.46; H, 4.99; N, 42.12.

Compound (2) (500 mg, 2.5 mmol), Bu4NI (95 mg, 0.25 mmol), THF (250 mL) and CH3CN (250 mL) were placed into a dry 1-liter, three-necked flask equipped with dropping funnel, a gas inlet tube, a gas outlet tube, reflux condenser and mechanical stirrer, and maintained under a positive pressure of nitrogen. The reaction mixture was heated at 60° C. for 20 mm and then cooled to room temperature. H5I06 (1.14 g, 5 mmol) was added and the reaction mixture was stirred vigorously at room temperature for 1 hour. The resulting solution was treated with saturated aqueous Na2S2O3 (5 mL) and was then concentrated under reduced pressure to dryness. The residue was treated with CH3CN (200 mL) and was filtered. The filtrate was concentrated and chromatographed on a column of silica gel (1.5% to 2% AcOH/EtOAc) to afford temozolomide (1) (280 mg).
1H NMR (400 MHz, DMSO-d6, δ): 8.80 (s, 1H), 7.80 (bs, 1H), 7.66 (bs, 1H), 3.43 (s, 3H).
…………………….
EXAMPLES
Example 1:
- Preparation of 3-Methyl-8-aminocarbonyl-imidazo[5,1-d]-1,2,3,5-tetrazin-4(3H)-one (Temozolomide).
-
Glacial acetic acid (25 ml), water (250 ml) and LiCl (225 g) were charged and the contents were stirred for 30 minutes and cooled to room temperature. 5-Amino-1-(N-methylcarbamoyl) imidazole-4-carboxamide (II) (25 g) was added and stirred the contents for further 30 minutes. The reaction mixture was cooled to 0°C and then added drop wise to NaNO2 solution (12.5 g in 50 ml water) at -10 to 5 °C. The reaction mass was stirred for 1 hr at 0-5 °C and then at room temperature for 5 hrs. To this reaction mixture, sodium thiosulphate solution (25 g in 250 ml of water) was added slowly and stirred for 20 minutes (solution A). This process yielded an acidic solution containing temozolomide.
……………………..
SYNTHESIS
EXAMPLES Example 1
A 250 ml reaction vessel equipped with a magnetic stirrer and a reflux condenser was charged with 8-cyano-3-methyl-[3H]-imidazo-[5,1-d]-tetrazin-4-one (10 grams, 0.0568 mol) and hydrochloric acid (36.5-38%, 50 ml). The reaction mixture was heated to 32-35° C. and stirring was maintained at this temperature for about 3 hours. A sample was withdrawn and analyzed by HPLC to verify that the high conversion was received. (If the content of the starting material 8-cyano-3-methyl-[3H]-imidazo-[5,1-d]-tetrazin-4-one is more than 2.5% by area according to HPLC, the stirring may be continued for additional one hour).
The reaction mixture was then cooled to 20° C. and 50 ml of acetone were added drop-wise while maintaining the temperature at 20° C. Stirring was continued for 15-30 minutes. The precipitated white crystals were washed with cold acetone (20 ml) and dried at 40° C. in vacuum to obtain 11.7 grams (0.0507 mol) of Temozolomide hydrochloride (89.3% yield). Purity (by HPLC): 99.6%.
…………………………
SYNTHESIS
EXAMPLES
The following Examples illustrate but do not in any way limit the present invention. Chemicals obtained from Aldrich Chemical Company (Milwaukee, Wis.) are identified by their catalog number. It should be noted that nomenclature may differ slightly between this specification and the Aldrich catalog.
Example 1 Preparation of Temozolomide (1)
Step A Preparation Compound (3)

5-Amino-1H-imidazole-4-carboxamide.HCl (4) (25 g, 0.154 mol) (Aldrich 16,496-8), CH2Cl2(0.6 L) and Et3N (45 mL) (Aldrich, 13,206-3) were placed into a dry 2-liter, three-necked flask equipped with dropping funnel, a gas inlet tube, a gas outlet tube, reflux condenser and mechanical stirrer, and maintained under a positive pressure of nitrogen at ambient temperature. The mixture was stirred, and a solution of 400 mL of 4-nitrophenyl chloroformate (34 g, 0.169 mol) (Aldrich, 16,021-0) in CH2Cl2was added dropwise. The reaction mixture was stirred vigorously for 4 hours and then left to stand for 18 hours at room temperature. The precipitate was collected by vacuum filtration and washed with H2O (1.5 L) to afford the product (3) as a pale yellow solid (42 g, 0.144 mol).
1H NMR (400 MHz, DMSO-d6, δ): 8.40 (d, 2H), 7.83 (s, 1H), 7.74 (d, 2H), 7.08 (bs, 1H), 6.95 (bs, 1H), 6.52 (s, 2H).
Step B Preparation of Compound (2)

Compound (3) (42 g, 0.144 mol) and DMF (0.27 L) were placed into a dry 1-liter, three-necked flask equipped with dropping funnel, a gas inlet tube, a gas outlet tube, reflux condenser and mechanical stirrer, and maintained under a positive pressure of nitrogen. The reaction mixture was cooled to 0° C., and methylhydrazine (10 mL, 0.188 mol) (Aldrich, M5,000-1) was added dropwise. The reaction mixture was stirred vigorously for 1 hour at 0° C. and was then poured into EtOAc (2.1 L). The precipitate was collected by vacuum filtration and was dried under vacuum (20 mm Hg, room temperature, 18 hours) to afford (2) as a tan solid (27.1 g, 0.137 mol).
1H NMR (400 MHz, DMSO-d6, δ): 7.62 (s, 1H), 6.85 (bs, 1H), 6.75 (bs,1H), 6.00 (s, 2H), 5.10 (s, 2H), 3.15, s, 3H).mp: 188° C. (dec.). Analysis: Calcd for C6H10N6O2: C, 36.36; H, 5.09; N, 42.41. Found: C, 36.46; H, 4.99; N, 42.12.
Step C Preparation of Temozolomide (1)

Compound (2) (500 mg, 2.5 mmol), Bu4NI (95 mg, 0.25 mmol), THF (250 mL) and CH3CN (250 mL) were placed into a dry 1-liter, three-necked flask equipped with dropping funnel, a gas inlet tube, a gas outlet tube, reflux condenser and mechanical stirrer, and maintained under a positive pressure of nitrogen. The reaction mixture was heated at 60° C. for 20 mm and then cooled to room temperature. H5IO6 (1.14 g, 5 mmol) was added and the reaction mixture was stirred vigorously at room temperature for 1 hour. The resulting solution was treated with saturated aqueous Na2S2O3 (5 mL) and was then concentrated under reduced pressure to dryness. The residue was treated with CH3CN (200 mL) and was filtered. The filtrate was concentrated and chromatographed on a column of silica gel (1.5% to 2% AcOH/EtOAc) to afford temozolomide (1) (280 mg).
1H NMR (400 MHz, DMSO-d6, δ): 8.80 (s, 1H), 7.80 (bs, 1H), 7.66 (bs, 1H), 3.43 (s, 3H).

TEMOZOLOMIDE
References
- Malcolm Stevens – interview, Cancer Research UK impact & achievements page
- Newlands ES, Stevens MF, Wedge SR, Wheelhouse RT, Brock C (January 1997). “Temozolomide: a review of its discovery, chemical properties, pre-clinical development and clinical trials”. Cancer Treat. Rev. 23 (1): 35–61. doi:10.1016/S0305-7372(97)90019-0. PMID 9189180.
- Stevens MF, Hickman JA, Langdon SP, Chubb D, Vickers L, Stone R, Baig G, Goddard C, Gibson NW, Slack JA et al. (November 1987). “Antitumor activity and pharmacokinetics in mice of 8-carbamoyl-3-methyl-imidazo[5,1-d]-1,2,3,5-tetrazin-4(3H)-one (CCRG 81045; M & B 39831), a novel drug with potential as an alternative to dacarbazine”. Cancer Res. 47 (22): 5846–52.PMID 3664486.
- Jacinto, FV; Esteller, M (August 2007). “MGMT hypermethylation: a prognostic foe, a predictive friend.”. DNA Repair 6 (8): 1155–60. doi:10.1016/j.dnarep.2007.03.013. PMID 17482895.
- Hegi ME, R, Hau, Mirimanoff et al. (March 2005). “MGMT gene silencing and benefit from temozolomide in glioblastoma”. N. Engl. J. Med. 352 (10): 997–1003. doi:10.1056/NEJMoa043331.PMID 15758010. More than one of
|last1=and|author=specified (help) - National Cancer Institute Of Canada Clinical Trials, Group; Hegi, ME; Mason, WP; Van Den Bent, MJ; Taphoorn, MJ; Janzer, RC; Ludwin, SK; Allgeier, A et al. (May 2009). “Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial”. Lancet Oncology 10 (5): 459–466. doi:10.1016/S1470-2045(09)70025-7. PMID 19269895.
- Sitbon Sitruk, L.; Sanson, M.; Prades, M.; Lefebvre, G.; Schubert, B.; Poirot, C. (2010). “Chimiothérapie à gonadotoxicité inconnue et préservation de la fertilité : Exemple du témozolomide☆”.Gynécologie Obstétrique & Fertilité 38 (11): 660–662. doi:10.1016/j.gyobfe.2010.09.002. PMID 21030284. edit
- Gilbert MR (March 2006). “New treatments for malignant gliomas: careful evaluation and cautious optimism required”. Ann. Intern. Med. 144 (5): 371–3. PMID 16520480.
- Pyrko P, Schönthal AH, Hofman FM, Chen TC, Lee AS (October 2007). “The unfolded protein response regulator GRP78/BiP as a novel target for increasing chemosensitivity in malignant gliomas”.Cancer Res. 67 (20): 9809–16. doi:10.1158/0008-5472.CAN-07-0625. PMID 17942911.
- Sheehan J, Cifarelli C, Dassoulas K, Olson C, Rainey J, Han S (2010). “Trans-sodium crocetinate enhancing survival and glioma response on magnetic resonance imaging to radiation and temozolomide”. Journal of Neurosurgery 113 (2): 234–239. doi:10.3171/2009.11.JNS091314. PMID 20001586.
- “Safety and Efficacy Study of Trans Sodium Crocetinate (TSC) With Concomitant Radiation Therapy and Temozolomide in Newly Diagnosed Glioblastoma (GBM)”. ClinicalTrials.gov. November 2011.
- Ueno T, Ko SH, Grubbs E et al. (March 2006). “Modulation of chemotherapy resistance in regional therapy: a novel therapeutic approach to advanced extremity melanoma using intra-arterial temozolomide in combination with systemic O6-benzylguanine”. Mol. Cancer Ther. 5 (3): 732–8. doi:10.1158/1535-7163.MCT-05-0098. PMID 16546988.
- Friedman, HS; Jiang, SX; Reardon, DA; Desjardins, A; Vredenburgh, JJ; Rich, JN; Gururangan, S; Friedman, AH et al. (March 2009). “Phase II trial of temozolomide plus o6-benzylguanine in adults with recurrent, temozolomide-resistant malignant glioma”. J. Clin. Oncol. 27 (8): 1262–7. doi:10.1200/JCO.2008.18.8417. PMC 2667825. PMID 19204199.
- http://labs.fhcrc.org/kiem/Hans-Peter_Kiem.html
- Dall’oglio S, D’Amico A, Pioli F, Gabbani M, Pasini F, Passarin MG, Talacchi A, Turazzi S, Maluta S (December 2008). “Dose-intensity temozolomide after concurrent chemoradiotherapy in operated high-grade gliomas”. J Neurooncol 90 (3): 315–9. doi:10.1007/s11060-008-9663-9. PMID 18688571.
- Osmani AH, Masood N; Masood (2012). “Temozolomide for relapsed primary CNS lymphoma”. J Coll Physicians Surg Pak 22 (9): 594–595. PMID 22980617.
- Chemotherapy Drug Shrinks Brain Tumors American Academy of Neurology, May 21, 2007
- Information for people undergoing treatment with temozolomide Cancer Research UK (CancerHelp UK)
Wang, et al., “Alternative Syntheses of the antitumor drug temozolomide avoiding the use of methyl isocyanates”, Journal of Chemical Society, Chemical Communication, Chemical Society, Letchworth, GB, p. 1687-1688 (1994).
Wang, et al., “Antitumor imidazotetrazines. Part 33. new syntheses of the antitumor drug temozolomide using ‘masked’ methyl isocyanates”, J. Chem. Soc., Perkin Trans. 1(21):2783-2787 (1995).
Wang, et al., “Synthetic studies of 8-carbamoylimidzo-‘5, 1-D!-1, 2, 3, 5-tetrazi n-4(3H)- one: a key derivative of antitumor drug temozolomide”, Bioorg. Med Chem. Lett., 6(2):185-188 (1996).
Yongfeng Wang, “A new route to the antitumor drug temozolomide, but not thiotemozolomide”, Chem. Commun., 4:363-364 (1997).
Wang, et al., “Antitumor Imidazotetrazines. 35. New Synthetic Routes to the Antitumor Drug Temozolomide”, J. org. Chem. 62(21):7228-7294 (1997).
Newlands, E.S., et al., “Temozolomide: a review of its discovery, chemical properties, pre-clinica development and clinical trials”, Cancer Treat. Rev. , 23(1):35-61 (1997).
Wang, et al., Antitumor Imidazotetrazines. Part 36. Conversion of 5-Amino-Imidazole-4-Carboxamide to . . . Journal of the Chemical Society, Perkin Transactions 1, Chemical Society, Letchworth, GB, 10:1669-1675 (1998).
1 Catapano CV, et al. Cancer Res. 1987, 47(18), 4884-4889.
[2] Sun S, et al. J Neurooncol. 2012.
[3] Bauer M, et al. PLoS One. 2012, 7(6):e39956.
[4] Wong ST, et al. Anticancer Res. 2012, 32(7), 2835-2841.
[5] Lin CJ, et al. PLoS One. 2012, 7(6), e38706.
[6] Gori JL, et al. Cancer Gene Ther. 2012.
| US5260291 | Oct 18, 1991 | Nov 9, 1993 | Cancer Research Campaign Technology Limited | Tetrazine derivatives |
| US20020133006 | Jan 16, 2002 | Sep 19, 2002 | Schering Corporation | Synthesis of temozolomide and analogs |
| US20050131227 | Jan 21, 2005 | Jun 16, 2005 | Schering Corporation | Synthesis of temozolomide and analogs |
| US20060183898 * | Feb 16, 2006 | Aug 17, 2006 | Olga Etlin | Process for preparing temozolomide |
| CN1487941A * | Jan 16, 2002 | Apr 7, 2004 | 先灵公司 | Synthesis of temozolomide and analogs |
| CN1706843A * | Apr 8, 2005 | Dec 14, 2005 | 江苏天士力帝益药业有限公司 | Temozolomide refining process |
| US20060183898 * | Feb 16, 2006 | Aug 17, 2006 | Olga Etlin | Process for preparing temozolomide |
| US20070225496 * | Mar 23, 2007 | Sep 27, 2007 | Palle Raghavendracharyulu Venk | rocess for preparing temozolomide |
| US8258294 * | Sep 28, 2007 | Sep 4, 2012 | Cipla Limited | Process for the preparation of temozolomide and analogs |
| EP2151442A2 | Jul 22, 2009 | Feb 10, 2010 | Chemi SPA | Process for preparing temozolomide |
| EP2374807A2 * | Sep 28, 2007 | Oct 12, 2011 | Cipla Limited | An improved process for the isolation of temozolomide |
| WO2008038031A1 | Sep 28, 2007 | Apr 3, 2008 | Cipla Ltd | An improved process for the preparation of temozolomide and analogs |
| WO2010140168A1 * | Jun 2, 2010 | Dec 9, 2010 | Ind-Swift Laboratories Limited | Improved process for preparing temozolomide |
| WO2011036676A2 | Sep 14, 2010 | Mar 31, 2011 | Ashwini Nangia | Stable cocrystals of temozolomide |
A REVIEW AND METHODS TO HANDLE PHOSGENE, TRIPHOSGENE SAFELY DURING DRUG SYNTHESIS


Phosgene
Phosgene is the chemical compound with the formula COCl2. This colorless gas gained infamy as a chemical weapon during World War I. It is also a valued industrial reagent and building block in synthesis of pharmaceuticals and other organic compounds. In low concentrations, its odor resembles freshly cut hay or grass.[3] In addition to its industrial production, small amounts occur naturally from the breakdown and the combustion oforganochlorine compounds, such as those used in refrigeration systems.[4] The chemical was named by combining the Greek words ‘phos’ (meaning light) and genesis (birth); it does not mean it contains any phosphorus (cf. phosphine).
 TRIPHOSGENE
TRIPHOSGENE
Triphosgene (bis(trichloromethyl) carbonate (BTC), C3Cl6O3) is a chemical compound that is used as a safer substitute for phosgene, because at room temperature it is a solid crystal, as opposed to phosgene which is a gas.Triphosgene crystals decompose above 200 °C READ …….http://www.buss-ct.com/e/company/publications/reaction_technology/eckert_reprint_CO6_2011-hr2.pdf
This compound is commercially available. It is prepared by exhaustive free radical chlorination of dimethyl carbonate:
- CH3OCO2CH3 + 3 Cl2 → CCl3OCO2CCl3 + 6 HCl
Triphosgene can be easily recrystallized from boiling hexanes to yield pure white crystals.
Triphosgene is used as a reagent in organic synthesis for a variety of chemical transformations including to bond one carbonyl group to two alcohols, and to convert an amine group into isocyanate.
The toxicity of triphosgene is the same as phosgene since it decomposes to phosgene on heating and upon reaction with nucleophiles. Even trace moisture leads to formation of phosgene. Therefore this reagent can be safely handled if one takes all the precautions as for phosgene.
Structure and basic properties
Phosgene is a planar molecule as predicted by VSEPR theory. The C=O distance is 1.18 Å, the C—Cl distance is 1.74 Å and the Cl—C—Cl angle is 111.8°.[5] It is one of the simplest acid chlorides, being formally derived from carbonic acid.
Industrially, phosgene is produced by passing purified carbon monoxide and chlorine gas through a bed of porous activated carbon, which serves as acatalyst:[4]
- CO + Cl2 → COCl2 (ΔHrxn = −107.6kJ/mol)
The reaction is exothermic, therefore the reactor must be cooled. Typically, the reaction is conducted between 50 and 150 °C. Above 200 °C, phosgene reverts to carbon monoxide and chlorine, Keq (300K) = 0.05. World production of this compound was estimated to be 2.74 million tonnes in 1989.[4]
Because of safety issues, phosgene is often produced and consumed within the same plant, and extraordinary measures are made to contain this toxic gas. It is listed on schedule 3 of the Chemical Weapons Convention: All production sites manufacturing more than 30 tonnes per year must be declared to the OPCW.[6] Although less dangerous than many other chemical weapons, such as sarin, phosgene is still regarded as a viablechemical warfare agent because it is so easy to manufacture when compared to the production requirements of more technically advanced chemical weapons such as the first-generation nerve agent tabun.[7]

Upon ultraviolet (UV) radiation in the presence of oxygen, chloroform slowly converts into phosgene by a radical reaction. To suppress thisphotodegradation, chloroform is often stored in brown-tinted glass containers. Chlorinated compounds used to remove oil from metals, such as automotive brake cleaners, are converted to phosgene by the UV rays of arc welding processes.[8]
Phosgene may also be produced during testing for leaks of older-style refrigerant gases. Chloromethanes (R12, R22 and others) were formerly leak-tested in situ by employing a small gas torch (propane, butane or propylene gas) with a sniffer tube and a copper reaction plate in the flame nozzle of the torch. If any refrigerant gas was leaking from a pipe or joint, the gas would be sucked into the flame via the sniffer tube and would cause a colour change of the gas flame to a bright greenish blue. In the process, phosgene gas would be created due to the thermal reaction. No valid statistics are available, but anecdotal reports suggest that numerous refrigeration technicians suffered the effects of phosgene poisoning due to their ignorance of the toxicity of phosgene, produced during such leak testing.[citation needed] Electronic sensing of refrigerant gases phased out the use of flame testing for leaks in the 1980s. Similarly, phosgene poisoning is a consideration for people fighting fires that are occurring in the vicinity of freon refrigeration equipment, smoking in the vicinity of a freon leak, or fighting fires using halon or halotron.
The great majority of phosgene is used in the production of isocyanates, the most important being toluene diisocyanate (TDI) and methylene diphenyl diisocyanate (MDI). These two isocyanates are precursors to polyurethanes.
Synthesis of carbonates
Significant amounts are also used in the production of polycarbonates by its reaction with bisphenol A.[4] Polycarbonates are an important class of engineering thermoplastic found, for example, in lenses in eye glasses. Diols react with phosgene to give either linear or cyclic carbonates (R = H, alkyl, aryl):
- HOCR2-X-CR2OH + COCl2 → 1/n [OCR2-X-CR2OC(O)-]n + 2 HCl
Synthesis of isocyanates
The synthesis of isocyanates from amines illustrates the electrophilic character of this reagent and its use in introducing the equivalent of “CO2+“:[9]
Such reactions are conducted in the presence of a base such as pyridine that absorbs the hydrogen chloride.
Laboratory uses
In the research laboratory phosgene still finds limited use in organic synthesis. A variety of substitutes have been developed, notably trichloromethyl chloroformate (“diphosgene“), a liquid at room temperature, and bis(trichloromethyl) carbonate (“triphosgene“), a crystalline substance.[10] Aside from the above reactions that are widely practiced industrially, phosgene is also used to produceacid chlorides and carbon dioxide from carboxylic acids:
- RCO2H + COCl2 → RC(O)Cl + HCl + CO2
Such acid chlorides react with amines and alcohols to give, respectively, amides and esters, which are commonly used intermediates. Thionyl chloride is more commonly and more safely employed for this application. A specific application for phosgene is the production of chloroformic esters:
- ROH + COCl2 → ROC(O)Cl + HCl
Although it is somewhat hydrophobic, phosgene reacts with water to release hydrogen chloride and carbon dioxide:
- COCl2 + H2O → CO2 + 2 HCl
Analogously, with ammonia, one obtains urea:
- COCl2 + 4 NH3 → CO(NH2)2 + 2 NH4Cl
Halide exchange with nitrogen trifluoride and aluminium tribromide gives COF2 and COBr2, respectively.[4]
History
Phosgene was synthesized by the British chemist John Davy (1790–1868) in 1812 by exposing a mixture of carbon monoxide and chlorine to sunlight. He named it “phosgene” in reference of the use of light to promote the reaction; from Greek, phos (light) and gene (born).[11] It gradually became important in the chemical industry as the 19th century progressed, particularly in dye manufacturing.
Further information: Use of poison gas in World War I and Second Italo-Abyssinian War
Following the extensive use of phosgene gas in combat during World War I, it was stockpiled by various countries as part of their secret chemical weapons programs.[12][13][14]
In May 1928, eleven tons of phosgene escaped from a war surplus store in central Hamburg.[15] 300 people were poisoned of whom 10 died.[15]
.

US Army phosgene identification poster from World War II
Phosgene was then only frequently used by the Imperial Japanese Army against the Chinese during the Second Sino-Japanese War.[16] Gas weapons, such as phosgene, were produced by Unit 731 and authorized by specific orders given by Hirohito (Emperor Showa) himself, transmitted by the chief of staff of the army. For example, the Emperor authorized the use of toxic gas on 375 separate occasions during the battle of Wuhan from August to October 1938.[17]
Phosgene is an insidious poison as the odor may not be noticed and symptoms may be slow to appear.[18] The odor detection threshold for phosgene is 0.4 ppm, four times the threshold limit value. Its high toxicity arises from the action of the phosgene on the proteins in the pulmonary alveoli, the site of gas exchange: their damage disrupts the blood-air barrier, causing suffocation. It reacts with the amines of the proteins, causing crosslinking by formation of urea-like linkages, in accord with the reactions discussed above. Phosgene detection badges are worn by those at risk of exposure.[4]
Sodium bicarbonate may be used to neutralise liquid spills of phosgene. Gaseous spills may be mitigated with ammonia.[19]
.
TRIPHOSGENE HANDLING
.

Left, reaction vessel with amino acid and triphosgene dissolved in THF; middle, appearance of the reaction mixture after addition of 2,4,6-collidine; and right, appearance of the reaction mixture after microwave irradiation.

Typical glassware standard equipment for the safety phosgenation with phosgene supply from triphosgene: (A) phosgene generator (V = 1 L, T = 85 °C) loaded with 600 g of triphosgene; (B) refluxer (water cooled, T = 15 °C); (C) phosgene line (Viton hose); (D) phosgenation reactor (V = 10 L, T = 110 °C); (E) refluxer (cryostat cooled, T = −30 °C); (F) off-gas line (Viton hose) from the top of the refluxer (E); (G) cooling trap (dry ice cooled, T = −60 °C); (H) off-gas line; (I) cryostat. The assembly of the equipment is somewhat reduced to effect more clarity of the ensemble.
.

.
Phosgene is quantitatively formed from solid triphosgene in a solvent-free and safe process without any reaction heat, catalyzed by planar N-heterocycles with deactivated imino functions.
The rate of phosgene generation is adjustable to the rate of phosgene consumption in the subsequent phosgenation reaction by thermal control, catalyst concentration, and in some cases, specific properties of selected metal phthalocyanines. A thermal runaway reaction of this process is impossible.
.
Use a safer process for generating phosgene.

Decomposition of triphosgene (1a) into carbon tetrachloride, carbon dioxide, and 1 equiv of phosgene (3)
Phosgene (COCl2) is useful in organic synthesis for chlorination, chlorocarbonylation, carbonylation, and dehydration; but its high toxicity discourages its use. Until now, the best substitute for COCl2 has been triphosgene [(CCl3O)2CO], a stable solid that has low vapor pressure. Although (CCl3O)2CO can be used in phosgenation reactions, removing the unreacted reagent from reaction mixtures is difficult because of its high boiling point. In contrast, COCl2 is easily removed by evaporating it.
(CCl3O)2CO reacts with silica gel, metal salts, or Lewis acids to generate 1 equiv of phosgene by an electrocyclic reaction. H. Eckert* and J. Auerweck at the University of Technology, Munich (Germany) report that pyridine and phthalocyanine derivatives catalyze the decomposition of (CCl3O)2CO to generate 3 equiv of COCl2.
The catalysts, phenanthridine , poly(2-vinylpyridine) , and phthalocyanines , convert liquid (CCl3O)2CO to the desired COCl2. The size and structure of the catalysts allow (CCl3O)2CO to react by the mechanism shown. The reaction was run at the 100-g scale to generate 22 L of gaseous COCl2 with an oil bath or an IR heater as the heat source. Because the catalysts are not soluble in (CCl3O)2CO, the process is considered to be heterogeneous catalysis.
.

Controlled transformation of triphosgene (1) into 3 equiv of phosgene (3) catalyzed by 4
Compounds 1 and 4a−4 h are commercially available products from Sigma-Aldrich, with the following purities: 1, 98% (IR νC═O 1820 cm−1, 13C NMR δ 108.0, 140.9); 4a, 98%; 4c, n.a.; 4d, 99%;4e, 97%; 4f, 97%; 4g, 90%; 4h, 85%.
Because the reaction is controlled by temperature, turning off the heat source causes the liquid (CCl3O)2CO to crystallize and stops the reaction, making the process safe. The reaction can be used to generate COCl2 externally or to produce it in situ. According to the authors, this method fulfills the goal of “safety phosgenation on demand of consumer”.
ORGANIC PROCESS RESEARCH AND DEVELOPMENT
A FRET approach towards potential detection of phosgene is presented, which is based on a selective chemical reaction between phosgene (or triphosgene as a simulant) and donor and acceptor fluorophores.

FRET has been applied in an experimental method for the detection of phosgene. In it, phosgene or rather triphosgene as a safe substitute serves as a linker between an acceptor and a donor coumarine (forming urea groups).[3] The presence of phosgene is detected at 5×10-5M with a typical FRET emission at 464 nm.


EXAMPLES OF USE OF TRIPHOSGENE
Chlorination of Aliphatic Primary Alcohols via Triphosgene-Triethylamine Activation
Caitlan E. Ayala, Andres Villalpando, Alex L. Nguyen, Gregory T. McCandless and Rendy Kartika*
*Department of Chemistry, 232 Choppin Hall, Louisiana State University, Baton Rouge, Louisiana 70803, United States, Email: rkartika lsu.edu
lsu.edu
C. E. Ayala, A. Villalpando, A. L. Nguyen, G. T. McCandless, R. Kartika, Org. Lett., 2012, 14, 3676-3679.
DOI: 10.1021/ol301520d (free Supporting Information)

Activation of primary aliphatic alcohols with triphosgene and triethylamine mixtures afforded either alkyl chloride or diethylcarbamate products, and the switch in selectivity appeared to be driven by sterics. The reaction conditions to achieve this highly useful transformation were unexceptionally mild and readily tolerated by a wide range of sensitive functionalities.

…………………………
.

.
ABACAVIR SULPHATE

VESTIPITANT
The following synthetic route was reported by Giuseppe Guercio et al from GlaxoSmithKline:
The initial chemical development synthetic route, derived from the one used by medicinal chemistry, involved several hazardous reagents, gave low yields and produced high levels of waste. Through a targeted process of research and development, application of novel techniques and extensive route scouting, a new synthetic route for GW597599 was developed. This paper reports the optimisation work of the third and last stage in the chemical synthesis of GW597599 and the development of a pilot-plant-suitable process for the manufacturing of optically pure arylpiperazine derivative 1. In particular, the process eliminated the use of triphosgene in the synthesis of an intermediate carbamoyl chloride, substantially enhancing safety, overall yield, and throughput.

source:
Org. Process Res. Dev., 2009, 13 (6), pp 1100–1110.
Org. Process Res. Dev., 2009, 13 (3), pp 489–493.
Org. Process Res. Dev., 2008, 12 (6), pp 1188–1194.
.
TIVOZANIB

VIAGRA

EFAVIRENZ ………EP2454244A1


Enantiomerically pure hydantoins are prepared from optically pure α-amino amides utilizing triphosgene. A mechanism for the racemization observed with 1,1′-carbonyldiimidazole (CDI) for this type of reaction is proposed.
D. Zhang, X. Xing, G. D. Cuny, J. Org. Chem., 2006, 71, 1750-1753.
Double acylation of a titanaselenide by triphosgene;
4,5-ethylenedithio-1,3-diselenol-2-one

References
- JMerck Index, 11th Edition, 7310.
- http://www.inchem.org/documents/icsc/icsc/eics0007.htm
- CBRNE – Lung-Damaging Agents, Phosgene May 27, 2009
- Wolfgang Schneider; Werner Diller (2005), “Phosgene”, Ullmann’s Encyclopedia of Industrial Chemistry, Weinheim: Wiley-VCH, doi:10.1002/14356007.a19_411
- Nakata, M.; Kohata, K.; Fukuyama, T.; Kuchitsu, K. (1980). “Molecular Structure of Phosgene as Studied by Gas Electron Diffraction and Microwave Spectroscopy. The rz Structure and Isotope Effect”.Journal of Molecular Spectroscopy 83: 105–117. doi:10.1016/0022-2852(80)90314-8.
- Annex on Implementation and Verification (“Verification Annex”)
- https://itportal.decc.gov.uk/cwc_files/S2AAD_guidance.pdf
- “Common Cleaners Can Turn Into Poison Gas”. American Iron Magazine. TAM Communications. Retrieved 14 October 2011.
- R. L. Shriner, W. H. Horne, and R. F. B. Cox (1943), “p-Nitrophenyl Isocyanate”, Org. Synth.; Coll. Vol. 2: 453
- Hamley, P. “Phosgene” Encyclopedia of Reagents for Organic Synthesis, 2001 John Wiley, New York. doi: 10.1002/047084289X.rp149
- John Davy (1812). “On a Gaseous Compound of Carbonic Oxide and Chlorine”. Philosophical Transactions of the Royal Society of London 102: 144–151. doi:10.1098/rstl.1812.0008.JSTOR 107310.
- Base’s phantom war reveals its secrets, Lithgow Mercury, 7/08/2008
- Chemical warfare left its legacy, Lithgow Mercury, 9/09/2008
- Chemical bombs sit metres from Lithgow families for 60 years, The Daily Telegraph, September 22, 2008
- Ryan, T.Anthony (1996). Phosgene and Related Carbonyl Halides. Elsevier. pp. 154–155. ISBN 0444824456.
- Yuki Tanaka, “Poison Gas, the Story Japan Would Like to Forget”, Bulletin of the Atomic Scientists, October 1988, p. 16–17
- Y. Yoshimi and S. Matsuno, Dokugasusen Kankei Shiryô II, Kaisetsu, Jugonen Sensô Gokuhi Shiryoshu, 1997, p. 27–29
- Borak J., Diller W. F. (2001). “Phosgene exposure: mechanisms of injury and treatment strategies”. Journal of Occupational and Environmental Medicine 43 (2): 110–9. doi:10.1097/00043764-200102000-00008. PMID 11227628.
- “Phosgene: Health and Safety Guide”. International Programme on Chemical Safety. 1998.
-
(a) Cotarca, L. and Eckert, H. Phosgenations − A Handbook; Wiley-VCH: Weinheim, 2003.
(b) Cotarca, L. and Eckert, H. Phosgenations − A Handbook; Wiley-VCH:Weinheim, 2003; pp 20− 21.
(c) Cotarca, L. and Eckert, H. Phosgenations − A Handbook;Wiley-VCH: Weinheim, 2003; pp 44− 520.
(d) Cotarca, L. and Eckert, H. Phosgenations − A Handbook; Wiley-VCH: Weinheim, 2003; p 41.(e) Cotarca, L. and Eckert, H.Phosgenations − A Handbook; Wiley-VCH: Weinheim, 2003; pp 14− 16, 613− 615.
-
Recent online information: www.ch.tum.de/oc1/HEckert/research.htm.
-
(a) Senet, J. P. The Recent Advance in Phosgene Chemistry; SNPE: Paris, 1997; Vol. 1.
(b) Pasquato, L.; Modena, G.; Cotarca, L.; Delogu, P.; Mantovani, S. J. Org. Chem. 2000, 65,8224– 8228
(d) Dunlap, K. L. In Kirk-Othmer Encyclopedia of Chemical Technology, 5 ed.;Wiley: New York, 2006; Vol. 18, pp 802− 814.(e) Nielsen, D. H.; Burke, T. G.; Woltz, P. J. H.; Jones, E. A. J. Chem. Phys. 1952, 20, 596– 604
(f) Gordon, E. P.;Enakaeva, V. G.; Korotchenko, A. V.; Mitrokhin, A. M. Russian Patent RU 2299852, 2007.
-
(a) Eckert, H.; Forster, B. Angew. Chem. 1987, 99, 922– 923 ; Angew. Chem., Int. Ed.,1987, 26, 894–895
(b) Eckert, H. TUM-Mitteilungen (Technische Universitaet Muenchen) 2006, 3, 68– 69(d) Triphosgene; Ubichem: U.K., 1999; CD-ROM.
-
(a) Eckert, H.; Drefs, N. Chemanager 2006, 3) 10
-
Eckert, H.; Dirsch, N.; Gruber, B. (former Dr. Eckert GmbH, now Buss Chem Tech AG) German Offen. DE 19740577, 1999 (Sep. 15, 1997), Chem. Abstr. 1999, 130, 211406.;
WO 9914159, 1999; Eur. Pat. EP 1017623, 2002; U.S. Patent US 6399822, 2002; Japanese Patent JP 2001516692, 2001.
-
Mole percent 4 referring to 3 phosgene equivalents of 1 .
-
(a) Leznoff, C. C.; Lever, A. B. P. Phthalocyanines, Properties and Applications; VCH:Weinheim, NY, 1989.
(b) Lever, A. B. P. Adv. Inorg. Chem. Radiochem. 1965, 7, 28– 114
(c) Ebert, N. A.; Gottlich, H. B. J. Am. Chem. Soc. 1952, 74, 2806
[ACS Full Text ], [CAS]
], [CAS] -
The weighing error of this procedure mainly comes from icy condensed humidity at the cool glassware of the cooling trap and is less than 0.5 g, determined by a series of weighings under the same conditions, the same equipment, temperature (T = −78 °C), and handling time <10 s, but without 3. Under these conditions evaporation of 3 (bp 8 °C) hardly ever happens and can be ignored.
-
Monitox plus gas monitor (COCl2) and phosgene badges from Compurhttp://www.compur.com/gasmessgeraete/front_content.php?idcat=7&changelang=3.
- Davy’s account of his discovery of phosgene
- International Chemical Safety Card 0007
- CDC – Phosgene – NIOSH Workplace Safety and Health Topic
- NIOSH Pocket Guide to Chemical Hazards
- U.S. CDC Emergency Preparedness & Response
- U.S. EPA Acute Exposure Guideline Levels
- Regime For Schedule 3 Chemicals And Facilities Related To Such Chemicals, OPCW website
- CBWInfo website
- Use of Phosgene in WWII and in modern-day warfare (Refer to Section 4.C of the article)
- An experience with accidental poisoning by heated tetrachlorethylene solvent
‘Female Viagra’ Flibanserin now on track for Q3 filing in USA

Flibanserin, girosa
167933-07-5 cas no
147359-76-0 (monoHCl)
- Bimt 17
- BIMT 17 BS
- Bimt-17
- Flibanserin
- Girosa
- UNII-37JK4STR6Z
Women with low libido in the US will have to wait even longer for approval of the first ever treatment for the condition after regulators requested more data on the forerunner flibanserin, delaying its submission until later this year.
The US Food and Drug Administration has asked manufacturer Sprout Pharmaceuticals for data on how flibanserin interacts with other medicines and also how it affects driving ability, after around 10% of patients experienced sleepiness while on the drug
Read more at: http://www.pharmatimes.com/Article/14-02-11/Female_Viagra_now_on_track_for_Q3_filing_in_USA.aspx#ixzz2tAWxwzRD
December 11, 2013 – Sprout Pharmaceuticals today announced that it has received and appealed the Food and Drug Administration’s (FDA) Complete Response Letter (CRL) for flibanserin through the Formal Dispute Resolution process.
Flibanserin is an investigational, once-daily treatment for Hypoactive Sexual Desire Disorder, or HSDD, in premenopausal women. HSDD is the most commonly reported form of female sexual dysfunction
read all here
A new drug being developed by Boehringer Ingelheim could give a boost to the sex drive of women with low libido. The drug, known as flibanserin, has been shown in clinical trials to increase their sexual desire when taken once a day at bedtime.
The results from four pivotal Phase III clinical trials on women with hypoactive sexual desire disorder (HSDD) were presented this week at the European Society for Sexual Medicine’s congress in Lyon, France. The trials showed that participants taking flibanserin had a significant improvement in their sexual desire compared to those given a placebo. They also experienced less of the distress associated with sexual dysfunction.
The drug was initially being investigated as a treatment for depression, and acts on the serotonin receptors in the brain – it is both a 5-HT1A receptor agonist and a 5-HT2A receptor antagonist. It is also a partial agonist at the dopamine D4 receptor.

Neurotransmitters such as serotonin are believed to be involved in sexual function, and antidepressants are commonly associated with a loss of libido, so this was an obvious side-effect to look out for during clinical trials in depression. But far from suppressing the libido in women, it appeared to have the opposite effect, so trials in women with HSDD were initiated.
Hormone replacement can improve the libido of women who have had their ovaries removed, but there is no available drug to treat those who have not. There have been accusations that pharma companies invent new diseases like HSDD in order to sell more medicines, but according to Kathleen Segraves, an assistant professor at Case Western Reserve University in the US who has worked in the field of sexual functioning for many years, this is not the case here. HSDD is a very real disorder, she says, and the potential for a treatment for these women is very exciting.

Flibanserin (code name BIMT-17; proposed trade name Girosa) is a drug that was investigated by Boehringer Ingelheim as a novel, non-hormonal treatment for pre-menopausal women with Hypoactive Sexual Desire Disorder (HSDD).[1][2] Development was terminated in October 2010 following a negative report by the U.S. Food and Drug Administration.[3]
HSDD is the most commonly reported female sexual complaint and characterized by a decrease in sexual desire that causes marked personal distress and/or personal difficulties. According to prevalence studies about 1 in 10 women reported low sexual desire with associated distress, which may be HSDD.[4] The neurobiological pathway of female sexual desire involves interactions among multiple neurotransmitters, sex hormones and various psychosocial factors. Sexual desire is modulated in distinct brain areas by a balance between inhibitory and excitatory neurotransmitters, serotonin acting as an inhibitor while dopamine and norepinephrine act as a stimulator of sexual desire.[5][6]Flibanserin is a 5-HT1A receptor agonist and 5-HT2A receptor antagonist that had initially been investigated as an antidepressant. Preclinical evidence suggested that flibanserin targets these receptors preferentially in selective brain areas and helps to restore a balance between these inhibitory and excitatory effects.[6] HSDD has been recognized as a distinct sexual function disorder for more than 30 years.
The proposed mechanism of action refers back to the Kinsey dual control model. Several sex steroids, neurotransmitters, and hormones have important excitatory or inhibitory effects on the sexual response. Among the neurotransmitters, the excitatory activity is driven by dopamine and norepinephrine, while the inhibitory activity is driven by serotonin. The balance between these systems is relevant for a healthy sexual response. By modulating these neurotransmitters in selective brain areas, flibanserin, a 5-HT1A receptoragonist and 5-HT2A receptor antagonist, is likely to restore the balance between these neurotransmitter systems.[6]
Several large pivotal Phase III studies with Flibanserin were conducted in the USA, Canada and Europe. They involved more than 5,000 pre-menopausal women with generalized acquired Hypoactive Sexual Desire Disorder (HSDD). The results of the Phase III North American Trials demonstrated that
Although the two North American trials that used the flibanserin 100 mg qhs dose showed a statistically significant difference between flibanserin and placebo for the endpoint of [satisfying sexual events], they both failed to demonstrate a statistically significant improvement on the co-primary endpoint of sexual desire. Therefore, neither study met the agreed-upon criteria for success in establishing the efficacy of flibanserin for the treatment of [Hypoactive Sexual Desire Disorder].
These data were first presented on November 16, 2009 at the congress of the European Society for Sexual Medicine in Lyon, France. The women receiving Flibanserin reported that the average number of times they had “satisfying sexual events” rose from 2.8 to 4.5 times a month. However, women receiving placebo reported also an increase of “satisfying sexual events” from 2.7 to 3.7 times a month.
Evaluation of the overall improvement of their condition and whether the benefit was meaningful to the women, showed a significantly higher rate of a meaningful benefit in the flibanserin-treated patient group versus the placebo group.The onset of the Flibanserin effect was seen from the first timepoint measured after 4 weeks of treatment and maintained throughout the treatment period.
The overall incidence of adverse events among women taking flibanserin was low, the majority of adverse events being mild to moderate and resolved during the treatment. The most commonly reported adverse events included dizziness, nausea, fatigue, somnolence and insomnia.

On June 18, 2010, a federal advisory panel to the U.S. Food and Drug Administration (FDA) unanimously voted against recommending approval of Flibanserin.
Earlier in the week, a FDA staff report also recommended non-approval of the drug. While the FDA still might approve Flibanserin, in the past, negative panel votes tended to cause the FDA not to approve.
On October 8, 2010, Boehringer Ingelheim announced that it would discontinue its development of flibanserin in light of the FDA advisory panel’s recommendation.
On June 27, 2013, Sprout Pharmaceuticals confirmed they had resubmitted flibanserin for FDA approval.
Flibanserin, chemically 1 -[2-(4-(3-trifluoromethylphenyl)piperazin-1 – yl)ethyl]-2,3-dihydro-1 H-benzimidazole-2-one was disclosed in form of its hydrochloride in European Patent No. 526,434 (‘434) and has the following chemical structure:

Process for preparation of flibanserin were disclosed in European Patent No. ‘434, U.S. Application Publication No. 2007/0032655 and Drugs of the future 1998, 23(1): 9-16.
According to European Patent No. ‘434 flibanserin is prepared by condensing 1-(2-chloroethyl)-2,3-dihydro-1 H-benzimidazol-one with m- trifluoromethyl phenyl piperazine. According to U.S. Application Publication No. 2007/0032655 flibanserin is prepared by condensing 1-[(3-trifluoromethyl)phenyl]-4-(2- chloroethyl)piperazine with 1 -(2-propenyl)-1 ,3-dihydro-benzimidazol-2H-one.
According to Drugs of the future 1998, 23(1): 9-16 flibanserin is prepared by reacting 1-(2-chloroethyl)-2,3-dihydro-1 H-benzimidazol-one with m- trifluoromethylphenylpiperazine.
PATENT
1-[2-(4-(3-trifluoromethyl-phenyl)piperazin-1-yl)ethyl]-2,3-dihydro-1H-benzimidazol-2-one
Compound 3
Hydrochloride salt (isopropanol) M.p. 230-231°C
Analysis

¹H NMR (DMSO-d₆/CDCL₃ 5:2) 11.09 (b, 1H), 11.04 (s, 1H), 7.5-6.9 (8H), 4.36 (t, 2H), 4.1-3.1 (10H)
CLIP

The compound 1-[2-(4-(3-trifluoromethyl-phenyl)piperazin-1-yl)ethyl]-2,3-dihydro-1 H- benzimidazol-2-one (flibanserin) is disclosed in form of its hydrochlorid in European Patent Application EP-A-526434 and has the following chemical structure:

Flibanserin shows affinity for the 5-HTιA and 5-HT2-receptor. It is therefore a promising therapeutic agent for the treatment of a variety of diseases, for instance depression, schizophrenia, Parkinson, anxiety, sleep disturbances, sexual and mental disorders and age associated memory impairment.

EXAMPLE……… EP1518858A1
375 kg of 1-[(3-trifluoromethyl)phenyl]-4-(2-cloroethyl)piperazin are charged in a reactor with 2500 kg of water and 200 kg of aqueous Sodium Hydroxide 45%. Under stirring 169.2 kg of 1-(2-propenyl)-1,3-dihydro-benzimidazol-2H-one, 780 kg of isopropanol, 2000 kg of water and 220 kg of aqueous Sodium Hydroxide 45% are added. The reaction mixture is heated to 75-85° C. and 160 kg of concentrated hydrochloric acid and 200 kg of water are added.
The reaction mixture is stirred at constant temperature for about 45 minutes. After distillation of a mixture of water and Isopropanol (about 3000 kg) the remaining residue is cooled to about 65-75° C. and the pH is adjusted to 6.5-7.5 by addition of 125 kg of aqueous Sodium Hydroxide 45%. After cooling to a temperature of 45-50° C., the pH value is adjusted to 8-9 by addition of about 4 kg of aqueous Sodium Hydroxide 45%. Subsequently the mixture is cooled to 30-35° C. and centrifuged. The residue thus obtained is washed with 340 l of water and 126 l of isopropanol and then with water until chlorides elimination.
The wet product is dried under vacuum at a temperature of about 45-55° C. which leads to 358 kg of crude flibanserin polymorph A. The crude product thus obtained is loaded in a reactor with 1750 kg of Acetone and the resulting mixture is heated under stirring until reflux. The obtained solution is filtered and the filtrate is concentrated by distillation. The temperature is maintained for about 1 hour 0-5° C., then the precipitate solid is isolated by filtration and dried at 55° C. for at least 12 hours.
The final yield is 280 kg of pure flibanserin polymorph A.
CLIP
Flibanserin may be prepared by reacting 1-(phenylvinyl)-2,3-dihydro-1H-benzimidazol-2-one (I) with 1,2-dichloroethane (II) in the presence of NaH in warm dimethylformamide. The resulting 1-(2-chloroethyl)-2,3-dihydro-1H-benzimidazol-one (III) is in turn coupled with commercially available m-trifluoromethylphenylpiperazine hydrochloride (IV) in the presence of sodium carbonate and catalytic potassium iodide in refluxing ethanol. The crude flibanserin hydrochloride (V) is then dissolved in aqueous ethanol and the pure base is precipitated upon addition of sodium hydroxide.
PICK UP INTERMEDIATES FROM CHEM24H.COM
1-(1-phenylvinyl)-1,3-dihydro-2H-benzimidazol-2-one (I)
1,2-dichloroethane (II)
1-(2-chloroethyl)-1,3-dihydro-2H-benzimidazol-2-one (III)
1-[3-(trifluoromethyl)phenyl]piperazine; N-[3-(trifluoromethyl)phenyl]piperazine (IV)
1-(2-[4-[3-(trifluoromethyl)phenyl]piperazino]ethyl)-1,3-dihydro-2H-benzimidazol-2-one (V)
PATENT


wherein R2 is as defined in formula X; with a compound of formula Xl:

According to another aspect of the present invention there is provided a novel compound or a salt thereof selected from the compounds of formula I, IV and VII:


Wherein R is hydrogen or an amino protecting group.
Preferable the amino protecting groups are selected from butyl, 1 ,1- diphenylmethyl, methoxymethyl, benzyloxymethyl, trichloroethoxymethyl, pyrrolidinomethyl, cyanomethyl, pivaloyloxymethyl, allyl, 2-propenyl, t- butyldimethylsilyl, methoxy, thiomethyl, phenylvinyl, 4-methoxyphenyl, benzyl, A- methoxybenzyl, 2,4-dimethoxybenzyl, 2-nitrobenzyl, t-butoxycarbonyl, benzyloxycarbonyl, phenoxycarbonyl, 4-chlorophenoxycarbonyl, A- nitrophenoxycarbonyl, methoxycarbonyl and ethoxycarbonyl. Still more preferable protecting groups are selected from t- butoxycarbonyl, ethoxycarbonyl, methoxycarbonyl, benzyloxycarbonyl, phenoxycarbonyl, phenylvinyl and 2-propenyl.
R1 is independently selected from chlorine, bromine, iodine, methanesulphonate, trifluoromethanesulphonate, paratoluenesulphonate or benzenesulphonate. Preferable R1 is independently selected from chlorine, bromine or iodine and more preferable R1 is chlorine.
Wherein R2 is hydrogen or an amino protecting group.
The amino protecting group may be any of the groups commonly used to protect the amino function such as alkyl, substituted alkyl, hetero substituted alkyl, substituted or unsubstituted unsaturated alkyl, alkyl substituted hetero atoms, substituted or unsubstituted phenyl, substituted or unsubstituted benzyl, alkyoxy carbonyl groups and aryloxy carbonyl groups.
Preferable the amino protecting groups are selected from butyl, 1 ,1 – diphenylmethyl, methoxymethyl, benzyloxymethyl, trichloroethoxymethyl, pyrrolidinomethyl, cyanomethyl, pivaloyloxymethyl, allyl, 2-propenyl, t- butyldimethylsilyl, methoxy, thiomethyl, phenylvinyl, 4-methoxyphenyl, benzyl, A- methoxybenzyl, 2,4-dimethoxybenzyl, 2-nitrobenzyl, t-butoxycarbonyl, benzyloxycarbonyl, phenoxycarbonyl, 4-chlorophenoxycarbonyl, A- nitrophenoxycarbonyl, methoxycarbonyl and ethoxycarbonyl. Still more preferable protecting groups are selected from t- butoxycarbonyl, ethoxycarbonyl, methoxycarbonyl, benzyloxycarbonyl, phenoxycarbonyl, phenylvinyl and 2-propenyl. The following examples are given for the purpose of illustrating the present invention and should not be considered as limitations on the scope and spirit of the invention.
EXAMPLES Example 1
A mixture of sodium hydroxide (47 gm) and i-(α-methylvinyl) benzimidazol-2-one (100 gm) in dimethylformamide (400 ml) was .stirred for 1 hour at room temperature. Dibromoethane (217 gm) was slowly added to the mixture and stirred at 1 hour 30 minutes. The resulting solution after addition water (500 ml) was extracted with ethyl acetate. The combined ethyl acetate extract washed with water. After drying the solvent was removed under vacuum to yield 132 gm of 1 ,3-dihydro-1-(2-bromoethyl)-3-isopropenyl-2H-benzimidazol- 2-one as a yellow oily liquid.
Example 2 A mixture of 1 ,3-dihydro-1-(2-bromoethyl)-3-isopropenyl-2H- benzimidazol-2-one (100 gm), diethanolamine (175 ml), sodium carbonate (40 gm) and potassium iodide (10 gm) was heated to 90 to 95 deg C and stirred for 2 hours. The reaction mass was cooled to room temperature and added water (500 ml). The resulting mixture extracted into ethyl acetate and the organic layer washed with water. After drying the solvent was removed under vacuum to yield 105 gm of 1 ,3-dihydro-1-[2-[N-bis-(2-hydroxyethyl)amino]ethyl]-3-isopropenyl- 2H-benzimidazol-2-one as a thick yellow oily liquid.
Example 3
To the mixture of 1 ,3-dihydro-1-[2-[N-bis-(2-hydroxyethyl)amino]ethyl]-3- isopropenyl-2H-benzimidazol-2-one (100 gm) obtained as in example 2 and chloroform (300 ml), thionyl chloride (95 ml) was slowly added. The mixture was heated to reflux and stirred for 2 hours. The excess thionyl chloride and chloroform was distilled off to yield 98 gm of 1 ,3-dihydro-1-[2-[N-[bis-(2- chloroethyl)amino]ethyl]-3-isopropenyl-2H-benzimidazol-2-one as a brown coloured sticky residue.
Example 4
1 ,3-dihydro-1-[2-[N-[bis-(2-chloroethyl)amino]ethyl]-3-isopropenyl-2H- benzimidazol-2-one (98 gm) obtained as in example 3 was added to water (500 ml) and concentrated hydrochloric acid (200 ml) mixture. The mixture was heated to 60 to 65 deg C and stirred for 1 hour. The contents of the flask cooled to room temperature and pH of the solution adjusted to 9 – 10 with 10% sodium hydroxide solution. The resulting solution extracted with ethyl acetate and washed the organic layer with water. Evaporate the solvent under reduced pressure to yield 82 gm of 1 ,3-dihydro-1-[2-[N-bis-(2-chloroethyl)amino]ethyl]- 2H-benzimidazol-2-one as a dark brown coloured oily liquid
Example 5
A mixture of 1 ,3-dihydro-1-[2-[N-bis-(2-chloroethyl)amino]ethyl]-1,2-H- benzimidazol-2-one (82 gm) obtained as in example 4, xylene (300 ml) and m- trifluoromethyl aniline (58 gm) was refluxed for 64 hours. The reaction mass was cooled to room temperature and filtered to obtain 1-[2-(4-(3- thfluoromethylphenyl)piperazin-1-yl)ethyl]-2,3-dihydro-1 H-benzimidazole-2-one hydrochloride (Flibanserin hydrochloride) as a light brown coloured solid.
The crude flibanserin hydrochloride was purified in isopropyl alcohol to give 85 gm of pure flibanserin hydrochloride as off white solid.
Example 6
Piperazine (12 gm), toluene(60 ml) and tetra butyl ammonium bromide (1 gm) mixture was heated to 60 deg C, added 1 ,3-dihydro-1-(2-bromoethyl)-3- isopropenyl-2H-benzimidazol-2-one (10 gm) and stirred for 4 hours at 90 to 95 deg C. The mixture was cooled to 60 deg C and added water (50 ml). The separated toluene layer distilled under vacuum to give 8.5 gm of 1 ,3-dihydro-1- (2-piperazinyl)ethyl-3-isopropenyl-2H-benzimidazol-2-one as a white solid.
Example 7
To the mixture of concentrated hydrochloric acid (20 ml) and water (100 ml) was added 1 ,3-dihydro-1-(2-piperazinylethyl)-3-isopropenyl-2H- benzimidazol-2-one (10 gm) obtained as in example 6 and heated to 60 to 65 deg C 1 hour. The mixture was cooled to room temperature and pH of the solution was adjusted to 9 – 10 with 10% sodium hydroxide solution, extracted with ethyl acetate and the organic layer was washed with water. After drying the solvent was removed under vacuum to yield 8.5 gm of 1 ,3-dihydro-1-(2- piperazinyl ethyl)-2H-benzimidazol-2-one as a white solid.
Example 8
3-trifluoromethylaniline (40 gm) and hydrobromic acid (85 ml; 48- 50%w/w) mixture was cooled to 0 to 5 deg C. To this mixture added sodium nitrite solution (18.5 gm in 25 ml of water) at 5 to 10 deg C and copper powder (1 gm). The temperature was slowly raised to 50 to 55 deg C and stirred for 30 minutes. Added water (200 ml) to reaction mass and applied steam distillation, collected m-trifluoromethylbromobenzene as oily liquid. The oily liquid washed with sulfuric acid for two times (2 X 10 ml) followed by washed with water (2 X 20 ml) and dried the liquid with sodium sulphate to give 22 gm of m- trifluoromethylbromobenzene.
Example 9
To a mixture of 1 ,3-dihydro-1-(2-piperazinyl ethyl)-2H-benzimidazol-2- one (10 gm) obtained as in example 7, m-trifluoromethylbromobenzene (9 gm) obtained as in example 8, sodium tert-butoxide (5.5 gm), palladium acetate (4.5 mg) and xylene (80 ml) was added tri-tert.-butylphosphine (0.2 ml). The mixture was heated to 120 deg C and stirred for 3 hours. The reaction mass was cooled, added water (100 ml) and extracted with ethyl acetate and the organic layer was washed with water. After drying the solvent was removed under vacuum to yield
10 gm of 1-[2-(4-(3-trifluoromethylphenyl)piperazin-1-yl)ethyl]-2,3-dihydro-1 H- benzimidazole-2-one (Flibanserin).
Example 10
To a mixture of 1 ,3-dihydro-1-[2-[N-bis-(2-hydroxyethyl)amino]ethyl]-3- isopropenyl-2H-benzimidazol-2-one (100 gm) obtained as in example 3, cyclohexane (400 ml) and sodium carbonate (35 gm) was added benzene sulfonyl chloride (116 gm) at room temperature. The mixture was heated to 80 to
85 deg C and stirred for 8 hours . The contents were cooled to room temperature and added water (500 ml). Distilled the organic layer to give 182 gm of 1 ,3-dihydro-1-[2-[N-[bis-(2-benzenesulfonyloxy)- ethyl]amino]ethyl]-3- isopropenyl- 2H-benzimidazol-2-one.
Example 11
1 ,3-dihydro-1 -[2-[N-[bis-(2-benzenesulfonyloxy)- ethyl]amino]ethyl]-3- isopropenyl- 2H-benzitηidazol-2-one (100 gm) obtained as in example 10, dimethylformamide (500 ml) and sodium corbonate (18 gm) was mixed and heated to 70 deg C. To the mixture was added m-trifluoromethyl aniline (27 gm) and heated to 80 to 85 deg C, stirred for 5 hours. The reaction mass was cooled and added water (2000 ml), filtered the solid to yield 1 ,3-dihydro-1-[2-[4-(3- trifluoromethylphenyl)piperazinyl]ethyl]-3-isopropenyl-2H benzimidazol-2-one. Example 12
1 ,3-dihydro-1-[2-[N-[bis-(2-benzenesulfonyloxy)- ethyl]amino]ethyl]-3- isopropenyl- 2H-benzimidazol-2-one (100 gm) obtained as in example 11 added to the mixture of water (500 ml) and concentrated hydrochloric acid (200 ml), heated to 65 deg C and stirred for 1 hour. The reaction mass was cooled to room temperature and pH adjusted to 10 to 10-5 with 10% sodium hydroxide solution. The resulting mixture was extracted with ethyl acetate and the organic
■ layer was washed with water. After drying the solvent was removed under vacuum to yield 87 gm of 1-[2-(4-(3-trifluoromethylphenyl)piperazin-1-yl)ethyl]- 2,3-dihydro-1 H-benzimidazole -2-one (Flibanserin).
Paper
Journal of Pharmaceutical and Biomedical Analysis, v.57, 2012 Jan 5, p.104(5)
Isolation and structural elucidation of flibanserin as an adulterant in a health supplement used for female sexual performance enhancement
Low, Min-Yong et al
http://www.sciencedirect.com/science/article/pii/S0731708511004833

This proposed formula and structure was further confirmed by 1H and 13C NMR data which indicated the presence of 20 carbon atoms and 21 protons.
1H NMR
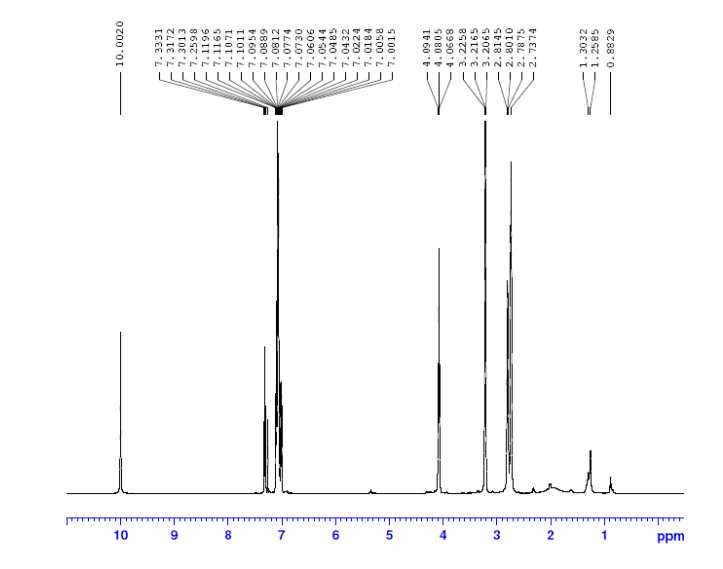
13C NMR
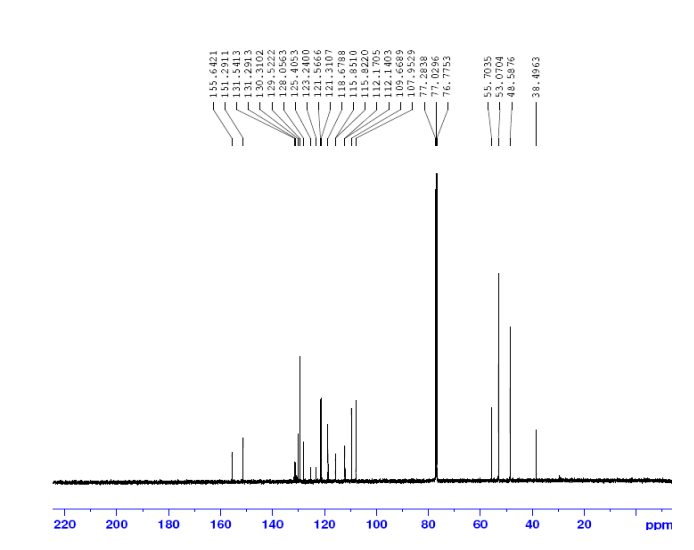
1D and 2DNMR data were used to assign the protons and carbon atoms.
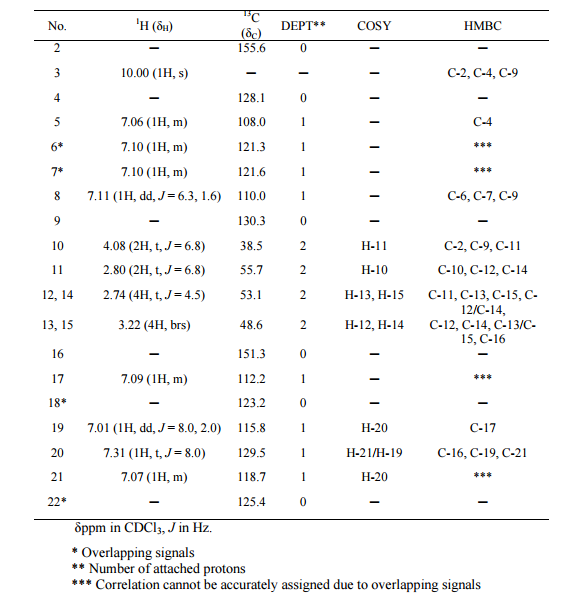
In the1H NMR spectrum , a sharp singlet at 10.00 ppm integrating for one
proton is a typical proton attached to nitrogen. HMBC correlated this proton to C-2, C-4, and C-9 suggesting that it was H-3.
Complex signals were observedbetween 7.00 to 7.31 ppm, integrating for eight protons. A triplet at 7.31 ppm,integrating for a proton has a coupling constant of 8.0 Hz. HMBC correlated thisproton with C-16, C-19, and C-21 suggesting that it was H-20.
A double-doubletsplitting pattern at chemical shift 7.11 ppm, integrating for a proton, has couplingconstants of 6.3 Hz and 1.6 Hz.
HMBC correlated this proton to C-6, C-7, and C-9 showing that it was H-8. Overlapped signals were observed from 7.04 ppm to7.10 ppm, integrating for five protons. A double-doublet splitting pattern at 7.01ppm with coupling constant 8.0 Hz and 2.0 Hz, integrating for a proton was
observed.
HMBC correlated this proton to C-17 suggesting that it was either H-19or H-21. Four triplet signals were also observed from 2.73 ppm to 4.08 ppm,integrating for a total of twelve protons.
Two of these triplet signals at 2.74 ppmand 3.22 ppm integrated for four protons each, suggesting overlapping signals ofmethylene protons. This was further confirmed by 13C and DEPT NMR.
13C and DEPT NMR data showed the signals of four methylene, eight methineand six quaternary carbon atoms. The DEPT signals at 53.1 ppm and 48.6 ppmhave intensities which were double of those from the rest of the methylene carbonsignals, suggesting two methylene carbon atoms each contributing to the signal at 53.1 ppm and 48.6 ppm.
DEPT

HMQC results further indicated that these two methylene carbon signals at 53.1 ppm and 48.6 ppm were correlated to the protons signal at 2.73 ppm and 4.08 ppm respectively, which corresponded to four protons each. The finding confirmed overlapping methylene carbon signals (at 53.1 ppm and 48.6 ppm) and methylene proton signals (at 2.73 ppm and 4.08 ppm). Hence, the unknown compound has six methylene carbon atoms with a total of twelve methylene protons.
The chemical shifts of the twelve methylene protons suggested that they were attached to relatively electronegative atoms. It was speculated that the six methylene groups were attached to the nitrogen atoms and the electron withdrawing effect of these electronegative nitrogen atoms resulted in the deshielding of the protons. HMBC and COSY correlations were used to assign the rest of the protons
HMBC

HMQC
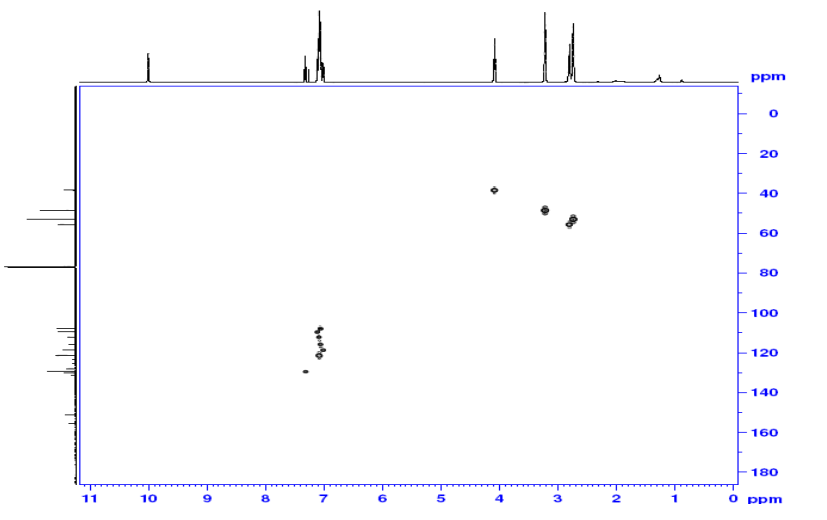
COSY
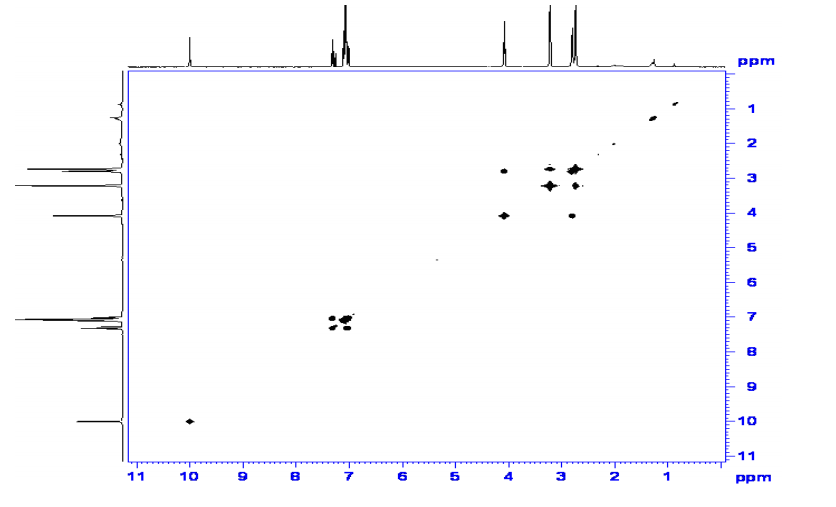
The 13C NMR data showed that there were two quaternary carbon at
155.6 ppm and 151.3 ppm. The carbon with chemical shift 155.6 ppm was C-2. Inthe structure of imidazolone, carbonyl carbon C-2 was attached to two nitrogenatoms which helped to withdraw electrons from oxygen to C-2. Hence, C-2 wasless deshielded as compared to a normal carbonyl carbon which has chemical shiftabove 170 ppm.
Eight methine carbons and two quaternary carbons with chemicalshifts above 108 ppm suggested the presence of two aromatic rings. Thequaternary carbon with chemical shift 125.4 ppm was C-22 which was attached tothree fluorine atoms. Due to the strong electron withdrawing effect of the fluorineatoms, C-22 was highly deshielded and had a high chemical shift.
The IR spectrum of the isolated compound showed absorption bands of amide (νC=O 1685 cm-1, νN-H (stretch) 3180 cm-1, νN-H (bending) 1610 cm-1), alkyl fluoride (νC-F1077 cm-1, 1112 cm-1, 1158 cm-1), aromatic ring (ν Ar-H 3028 cm-1, 3078 cm-1 andνC=C 1401 cm-1, 1446 cm-1, 1453 cm-1, 1468 cm-1, 1487 cm-1) and alkane (νC-H2891 cm-1, 2930 cm-1 2948 cm-).
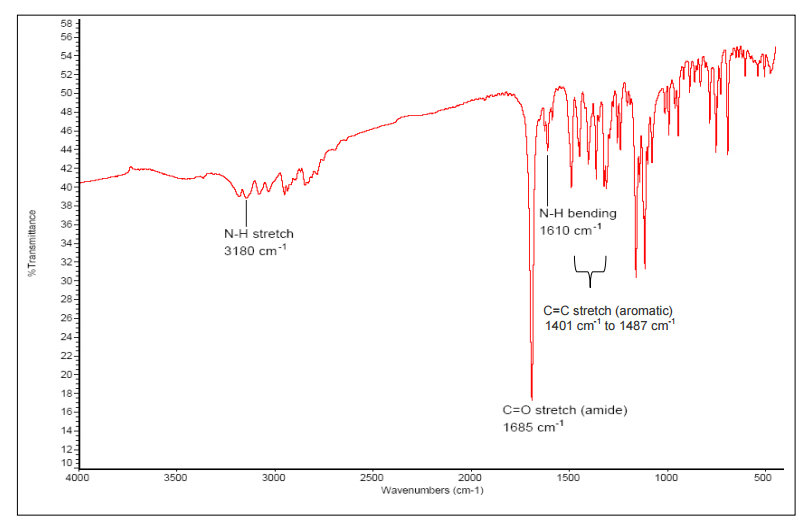
FOR MASS, HMBC ETC SEE………http://orgspectroscopyint.blogspot.in/2015/06/flibanserin.html
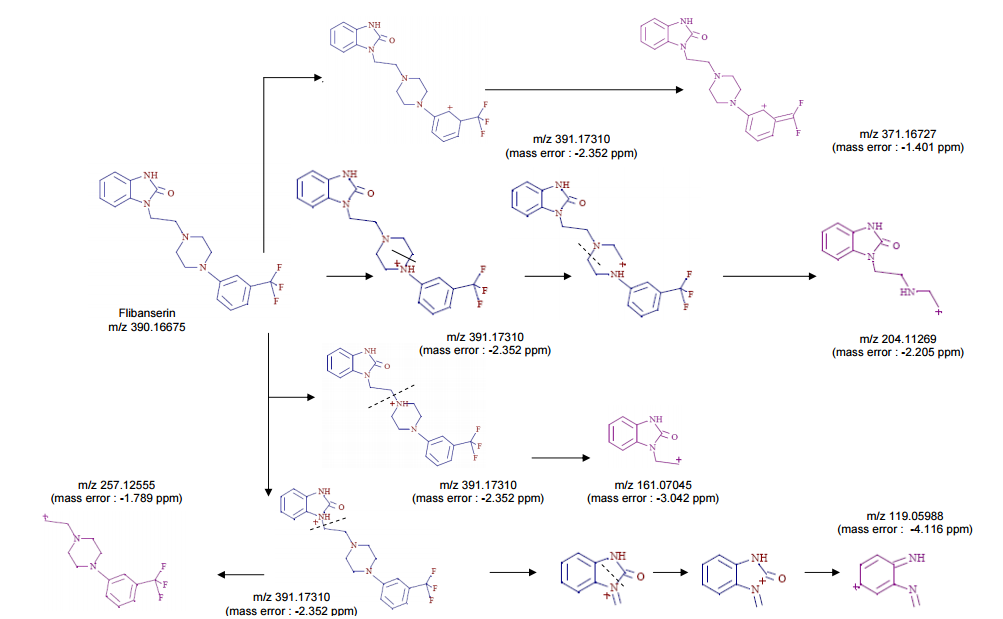
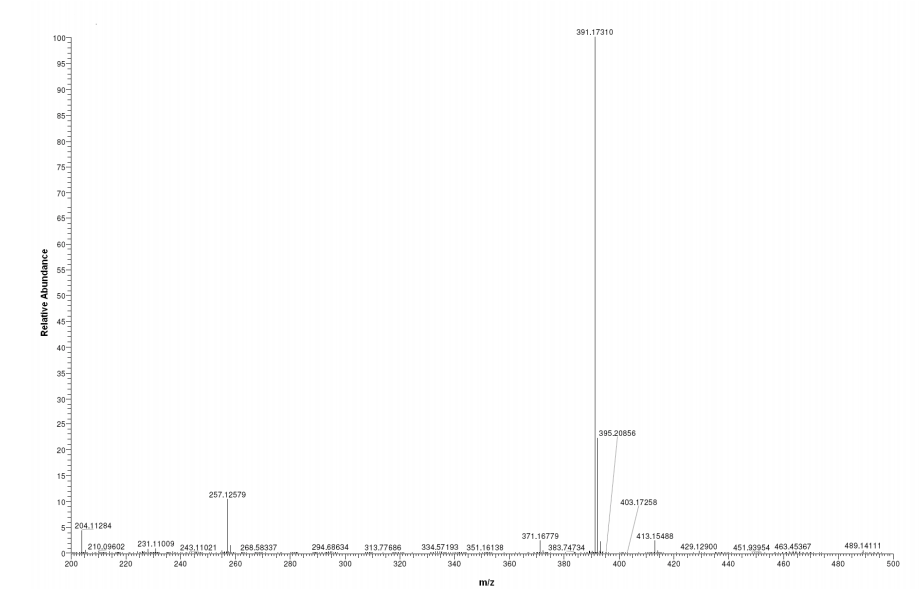
NMR PREDICT
1H NMR PREDICT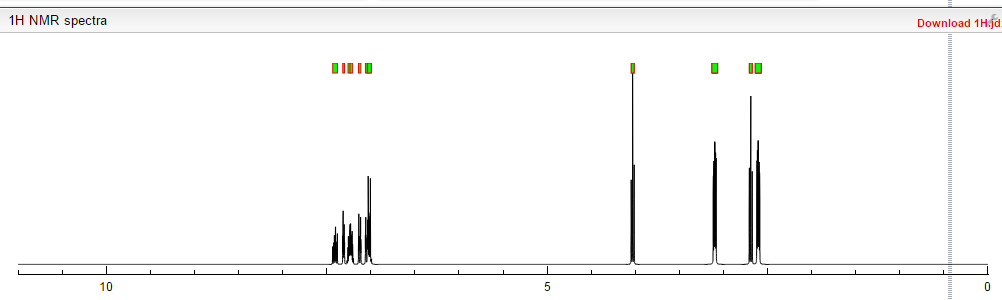
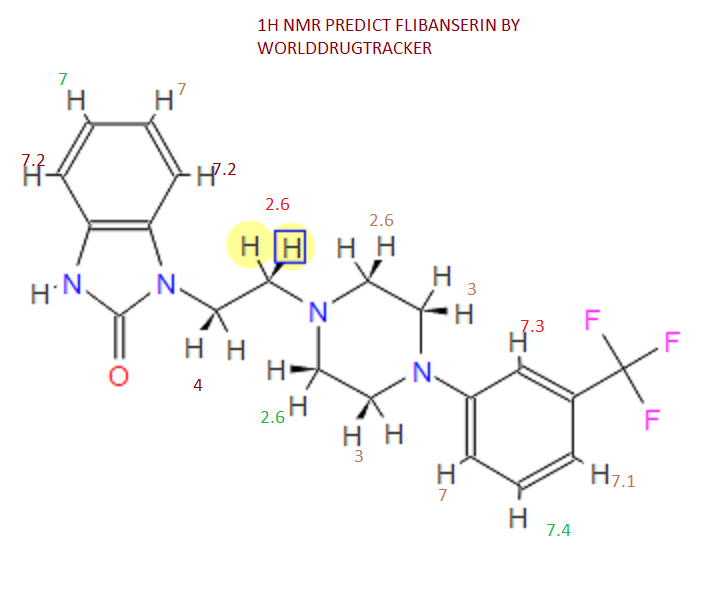
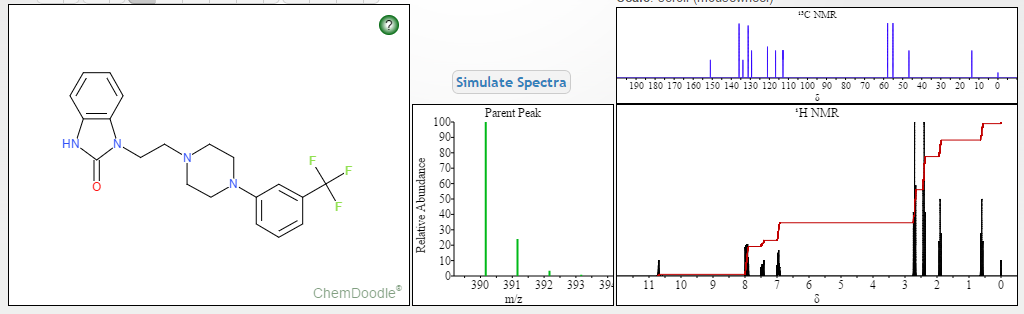
13C NMR PREDICT
COSY PREDICT
NMR PREDICT FROM MOLBASE
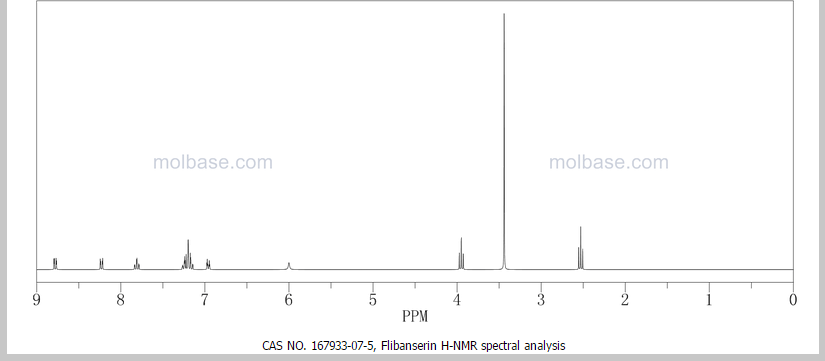
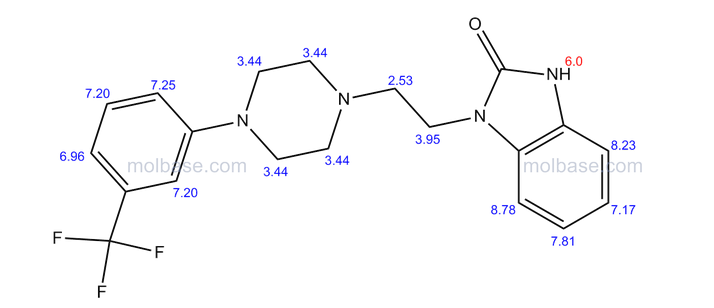
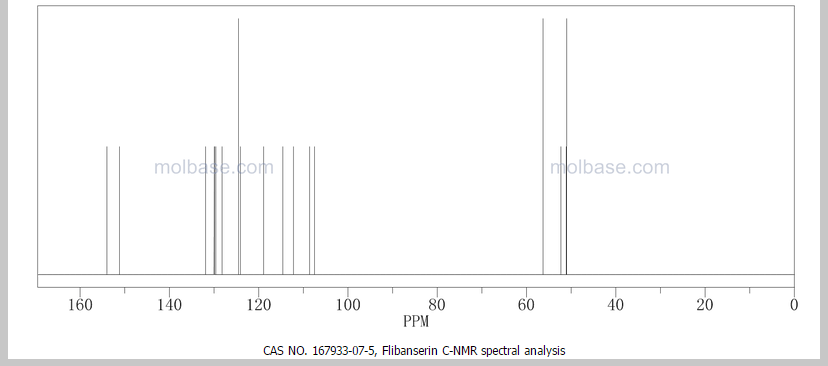

PATENT
US5576318, 1996
1 H NMR (DMSO-d6 /CDCL3 5:2) 11.09 (b, 1H), 11.04 (s, 1H), 7.5-6.9 (SH), 4.36 (t, 2H), 4.1-3.1 (10 H)
UPDATES………..
A Facile Route of Synthesis for Making Flibanserin

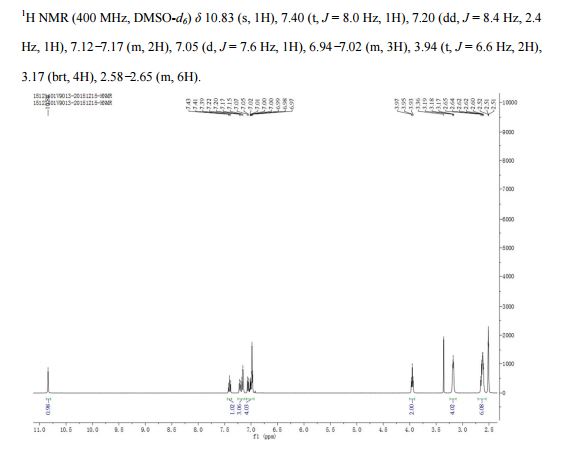
REFERENCES
- Borsini F, Evans K, Jason K, Rohde F, Alexander B, Pollentier S (summer 2002). “Pharmacology of flibanserin”. CNS Drug Rev. 8 (2): 117–142. doi:10.1111/j.1527-3458.2002.tb00219.x. PMID 12177684.
- Jolly E, Clayton A, Thorp J, Lewis-D’Agostino D, Wunderlich G, Lesko L (April 2008). “Design of Phase III pivotal trials of flibanserin in female Hypoactive Sexual Desire Disorder (HSDD)”. Sexologies 17 (Suppl 1): S133–4. doi:10.1016/S1158-1360(08)72886-X.
- Spiegel online: Pharmakonzern stoppt Lustpille für die Frau, 8 October 2010 (in German)
- Nygaard I (November 2008). “Sexual dysfunction prevalence rates: marketing or real?”. Obstet Gynecol 112 (5): 968–9.doi:10.1097/01.AOG.0000335775.68187.b2. PMID 18978094.
- Clayton AH (July 2010). “The pathophysiology of hypoactive sexual desire disorder in women”. Int J Gynaecol Obstet 110 (1): 7–11.doi:10.1016/j.ijgo.2010.02.014. PMID 20434725.
- Pfaus JG (June 2009). “Pathways of sexual desire”. J Sex Med 6 (6): 1506–33. doi:10.1111/j.1743-6109.2009.01309.x.PMID 19453889.
- Yves Aubert, Thesis, Leiden University. (Dec 11, 2012) Sex, aggression and pair-bond: a study on the serotonergic regulation of female sexual function in the marmoset monkey
- Viagra for women?
- Marazziti D, Palego L, Giromella A, et al. (June 2002). “Region-dependent effects of flibanserin and buspirone on adenylyl cyclase activity in the human brain”. Int. J. Neuropsychopharmacol. 5 (2): 131–40. doi:10.1017/S1461145702002869.PMID 12135537.
- Podhorna J, Brown RE (June 2000). “Flibanserin has anxiolytic effects without locomotor side effects in the infant rat ultrasonic vocalization model of anxiety”. Br J Pharmacol 130 (4): 739–746. doi:10.1038/sj.bjp.0703364. PMC 1572126.PMID 10864879.
- Brambilla A, Baschirotto A, Grippa N, Borsini F (December 1999). “Effect of flibanserin (BIMT 17), fluoxetine, 8-OH-DPAT and buspirone on serotonin synthesis in rat brain”. Eur Neuropsychopharmacol 10 (1): 63–7. doi:10.1016/S0924-977X(99)00056-5.PMID 10647099.
| EP0200322A1 * | Mar 18, 1986 | Nov 5, 1986 | H. Lundbeck A/S | Heterocyclic compounds |
| BE904945A1 * | Title not available | |||
| GB2023594A * | Title not available | |||
| US3472854 * | May 29, 1967 | Oct 14, 1969 | Sterling Drug Inc | 1-((benzimidazolyl)-lower-alkyl)-4-substituted-piperazines |
| US4954503 * | Sep 11, 1989 | Sep 4, 1990 | Hoechst-Roussel Pharmaceuticals, Inc. | 3-(1-substituted-4-piperazinyl)-1H-indazoles |
update………..
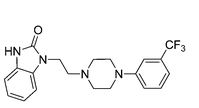
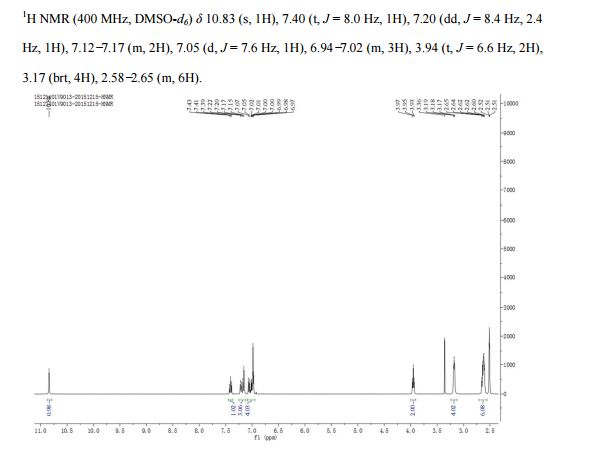
1-(2-(4-(3-(Trifluoromethyl)phenyl)piperazin-1-yl)ethyl)-1H-benzo[d]imidazol-2(3H)-one (1)

A novel and efficient route of synthesis for making flibanserin via 2-ethoxy-1H-benzo[d]imidazole (12) was described with excellent yield. This protocol provided a more facile approach toflibanserin.
A Facile Route of Synthesis for Making Flibanserin
http://pubs.acs.org/doi/abs/10.1021/acs.oprd.6b00108

aReagents and conditions: (a) ethyl benzoylacetate, 200 °C; (b) dichloroethane, NaH, DMF; (c) conc HCl (aq); (d) 1-(3-(trifluoromethyl)phenyl)piperazine hydrochloride, Na2CO3, KI, EtOH; (e)
- 3.Bietti, G.; Borsini, F.; Turconi, M.; Giraldo, E.; Bignotti, M. For treatment of central nervous system disorders. U.S. Patent 5,576,318, 1996.
- 4.Mohan Rao, D.; Krishna Reddy, P.; Venkat Reddy, B. Preparing benzoimidazol-2-one compound, useful to prepare flibanserin, comprises reacting benzoimidazol-2-one compound with 2-(2-hydroxy-ethylamino)-ethanol to give (bis-(hydroxy-ethyl)-amino)-ethyl-benzoimidazol-2-one compound. PCT. Int.WO2,010,128,516, 2010.5.
- 5.Vernin, G.; Domlog, H.; Siv, C.; Metzger, J.; El-Shafei, A. K.Synthesis of 1-alkyl and 1, 3-dialkyl-2-benzimidazolones from 1-alkenyl-2-benzimidazolones using phase-transfer catalysis technique J. Heterocycl. Chem. 1981, 18, 85– 89, DOI: 10.1002/jhet.5570180118
-
A patent application for the new synthetic route has been filed in China (CN201610527244.4).

aReagents and conditions: (a) ethyl acetoacetate, KOH, EtOH, xylene, reflux, 56%; (b) 1,2-dibromoethane, K2CO3, DMF, 50 °C, 50%; (c) K2CO3, CH3CN, 70 °C, 80%; (d) conc. HCl (aq), isopropanol, 70 °C; (e) NaOH (aq), rt, 72% over two steps.

aReagents and conditions: (a) tetraethyl orthocarbonate, AcOH, 70 °C, 94%; (b) 1-bromo-2-chloroethane, K2CO3, acetone, reflux, 75%; (c) K2CO3, NaI, H2O, reflux, 92%; (d) conc. HCl (aq), isopropanol, 70 °C; (e) NaOH (aq), 68% over two steps.
//////////////
Route to Benzimidazol-2-ones via Decarbonylative Ring Contraction of Quinoxalinediones: Application to the Synthesis of Flibanserin, A Drug for Treating Hypoactive Sexual Desire Disorder in Women and Marine Natural Product Hunanamycin Analogue
![]()


Route to Benzimidazol-2-ones via Decarbonylative Ring Contraction of Quinoxalinediones: Application to the Synthesis of Flibanserin, A Drug for Treating Hypoactive Sexual Desire Disorder in Women and Marine Natural Product Hunanamycin Analogue

INTRODUCTION
Benzimidazol-2-ones 1 are an important class of heterocycles and a privileged scaffold in medicinal chemistry. They consist of cyclic urea fused with the aromatic backbone, which can potentially interact in a biological system by various noncovalent interactions such as hydrogen bonding and π stacking. Benzimidazolone derivatives exhibit a wide range of biological activities, and they are useful in treating various diseases including cancer, type II diabetes, central nervous system disorders, pain management, and infectious disease.1 Selected compounds embedded with a benzimidazol-2-one moiety along with their use are captured in Figure 1. It is worth mentioning that oxatomide drug with a benzimidazol-2-one core was approved for marketing a few years ago.2a Very recently, US Food and Drug Administration approved a new drug called flibanserin for the treatment of hypoactive sexual desire disorder (HSDD) in females, which contains benzimidazol-2- one motif.2b
CONCLUSIONS
We have developed a mild and new protocol for the synthesis of benzimidazol-2-ones from quinoxalinediones through decarbonylation. The present methodology can be an addition to the toolbox to prepare benzimidazolones, and it will be useful in medicinal chemistry, particularly, late-stage functionalization of natural products, drug scaffolds, or an intermediate containing quinoxaline-2,3-diones. As direct application of this method, we have successfully developed a new route for the synthesis of recently approved drug flibanserin and a urea analogue of antibiotic natural product hunanamycin A. Later application demonstrates the utility of the present method in late-stage functionalization
Synthesis of 1-(2-(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)ethyl)-1,3-dihydro-2Hbenzo[d]imidazol-2-one (Flibanserin)
Flibanserin hydrochloride as white solid.
1H NMR (400MHz ,DMSO-d6) 11.06 (s, 1 H), 10.93 (br. s., 1 H), 7.54 – 7.41 (t, J = 7.9 Hz, 1 H), 7.36 – 7.22 (m, 3 H), 7.15 (d, J = 7.6 Hz, 1 H), 7.09 – 7.01 (m, 3 H), 4.30 (t, J = 6.7 Hz, 2 H), 4.01 (d, J = 11.6 Hz, 2 H), 3.75 (d, J = 10.4 Hz, 2 H), 3.54 – 3.43 (d, J = 4.2 Hz 2 H), 3.31 – 3.10 (m, 4 H);
HRMS (ESI): m/z calculated for C20H22ON4F3[M+H]+ 391.1740 found 391.1743;
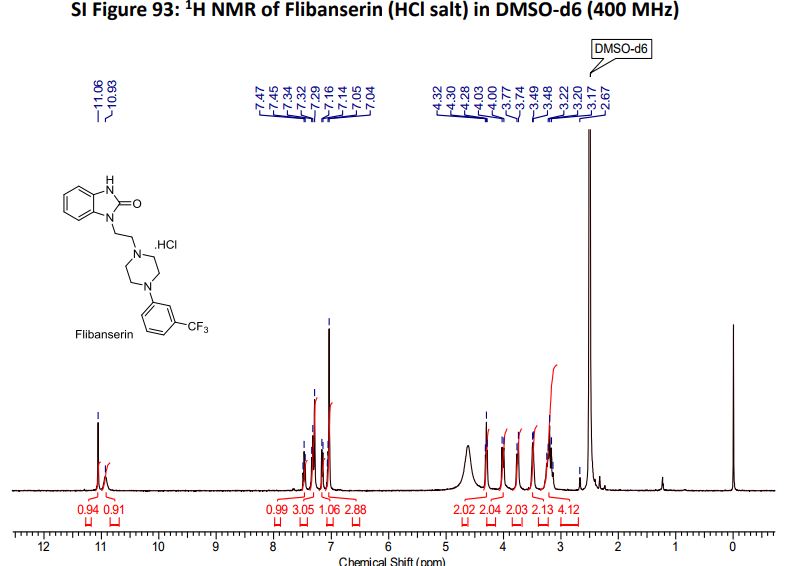
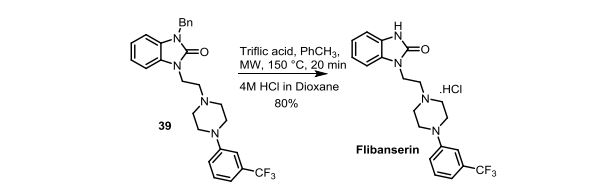

Scheme 4. Synthesis of Flibanserin through Ring Contraction
The same methodology was applied for the synthesis of flibanserin, also known as “female viagra”, which is the first approved medication for treating HSDD in women and is classified as a multifunctional serotonin agonist antagonist.(14, 15) Our synthesis of flibanserin commenced with 1-benzyl-1,4-dihydroquinoxaline-2,3-dione 36,(16) which was reacted with known chloride 37(17) under the basic condition in DMF to give the desired product 38 in good yield. Compound 38 was subjected for the decarbonylative cyclization under the optimized condition to afford the product 39 in 59% yield. Finally, the benzyl group was deprotected using trifluoromethanesulfonic acid in toluene under microwave irradiation,(8b, 18) which gave flibanserin in excellent yield (Scheme 4). The final product was isolated as HCl salt, and all of the spectral data are in agreement with the published data.(15c)

Rahul D. Shingare completed his M.Sc (Chemistry) from Fergusson College, Pune in 2008. He worked as a research associate in Ranbaxy and Lupin New drug discovery center, Gurgaon and Pune respectively until 2012 and currently pursuing his doctoral research in NCL – Pune from 2012.
Current Research Interests: Antibacterial Natural Product Hunanamycin A: Total Synthesis, SAR and Related Chemistry.
e-mail: rd.shingare@ncl.res.in

Akshay Kulkarni completed his M.Sc. from Ferguson College, Pune University in the year 2015 and joined our group as a Project Assistant in the month of October, 2015.
Current research interest: Synthesis of silicon incorporated biologically active antimalerial compounds.
e-mail : as.kulkarni@ncl.res.in
Dr.D. Srinivasa Reddy
Organic Chemistry Division
CSIR-National Chemical Laboratory
-
See, previous synthesis of Flibanserin:
(a) Bietti, G.; Borsini, F.; Turconi, M.; Giraldo, E.; Bignotti, M. For treatment of central nervous system disorders. U.S. Patent 5,576,318, 1996.
(b) Mohan, R. D.; Reddy, P. K.;Reddy, B. V. Process for the preparation of Flibanserin involving novel intermediates. WO2010128516 A2,2010.
(c) Yang, F.; Wu, C.; Li, Z.; Tian, G.; Wu, J.; Zhu, F.; Zhang, J.; He, Y.; Shen, J. A Facile route of synthesis for making Flibanserin Org. Process Res. Dev. 2016, 20, 1576 DOI: 10.1021/acs.oprd.6b00108
[ACS Full Text], [CAS] -
Xueong, X. Preparation method of Flibanserin. CN104926734 A, 2015.
//////////

AVASCULAR NECROSIS ; POST OPERATIVE AND POST SURGICAL COMPLICATIONS CASE ; AYURVEDA E.T.G AYURVEDASCAN DIAGNOSIS AND APPROACH
Recently a case of AVASCULAR NECROSIS , bilateral operated before one year, developed major complications in his both HIP-Joints severely. Surgeon, who have taken the case under his supervision, advised him to for HIP REPLACEMENT.
In this crisis satge, patient approched me and asked for the help for AYURVEDA TREATMENT.
HIS ETG AyurvedaScan traces are given below and some essential details are given below.




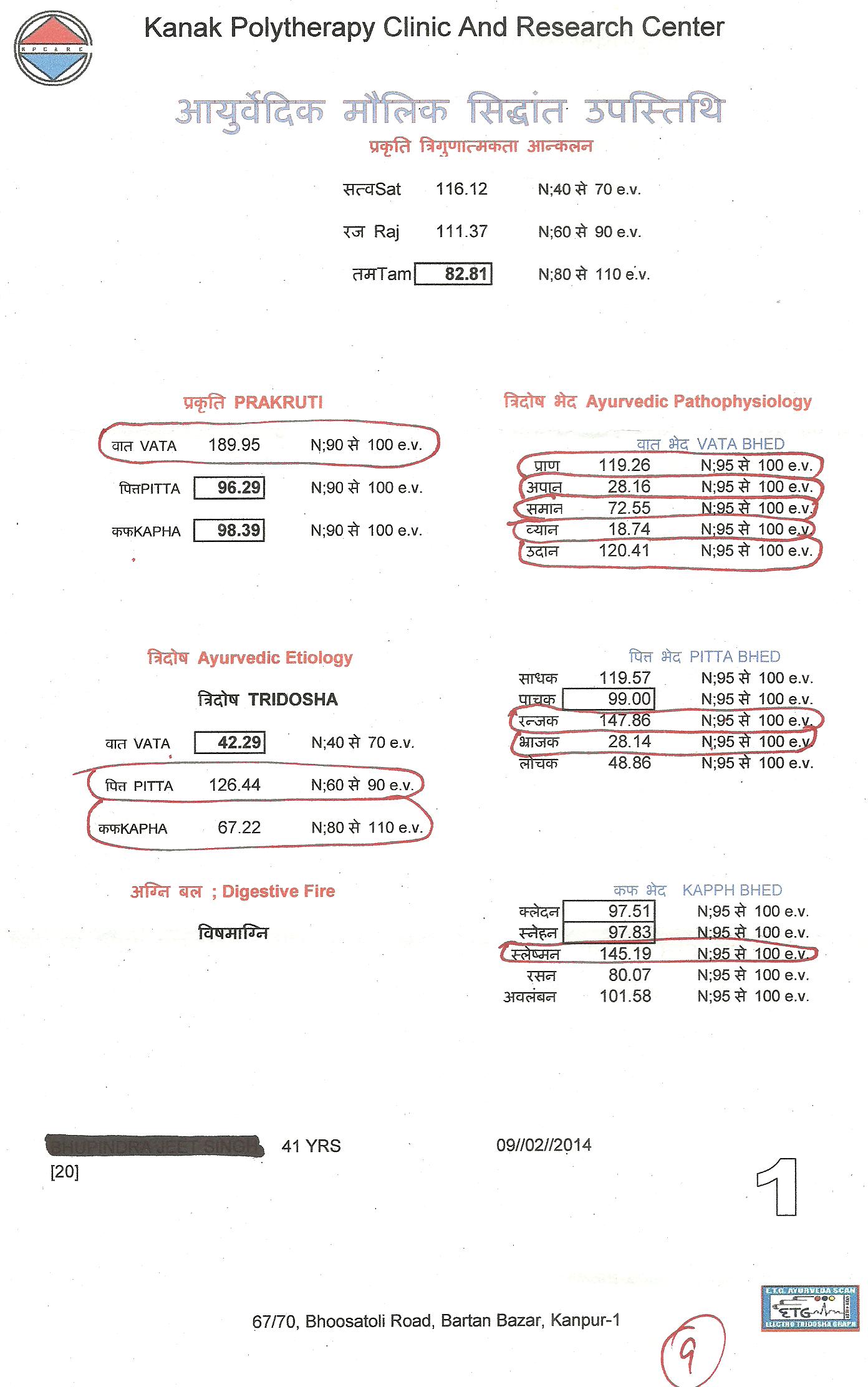
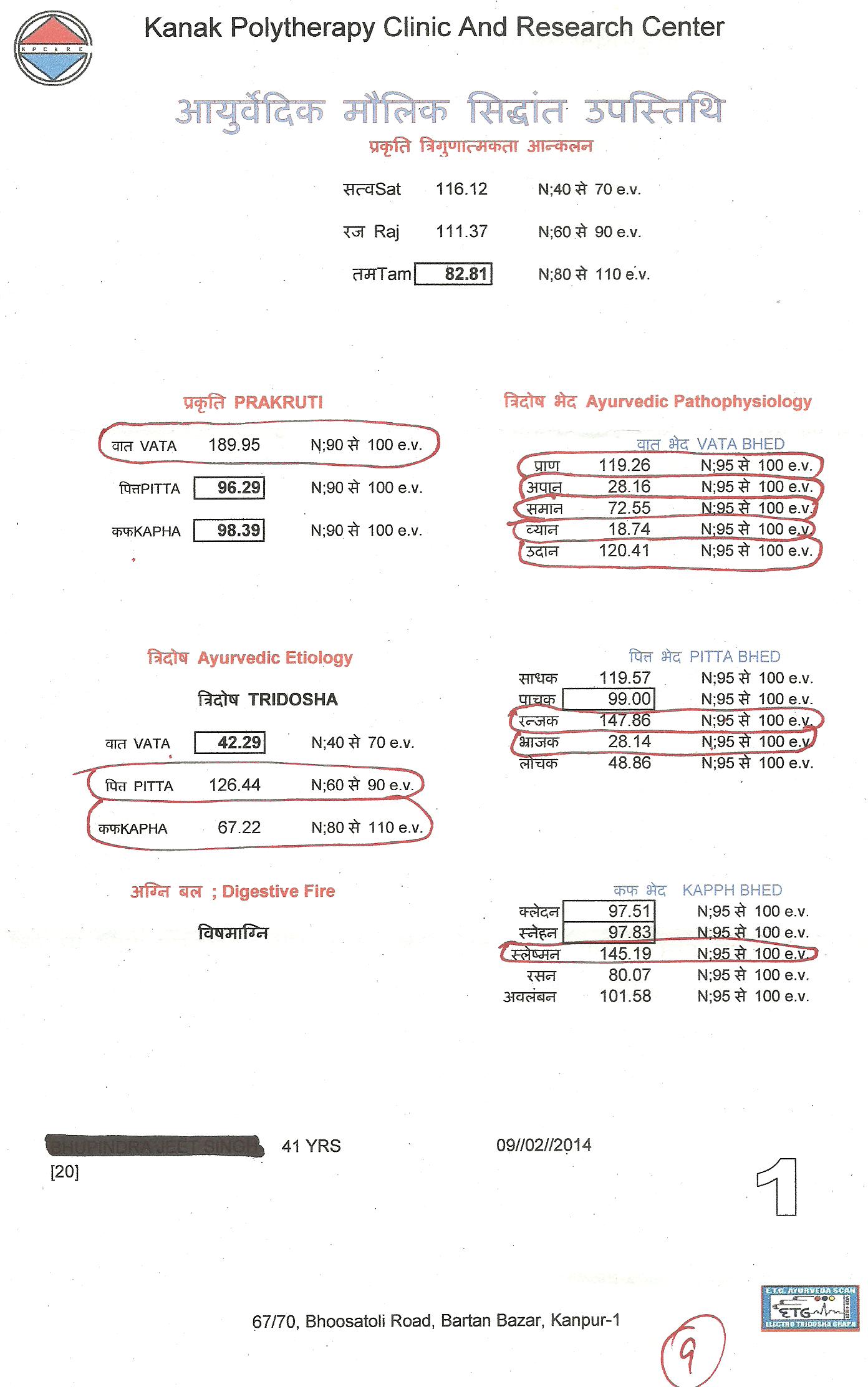

[TO BE LOADED SOON]
FDA Guidance on Polymorphic Compounds in Generic Drugs

The guidance issued by the US Food and Drug Administration advises companies on how to treat polymorphic drug compounds—those that exhibit multiple structural forms—in filing abbreviated new drug applications (ANDAs). The bottom line, according to the guidance, is that generic drug products containing the polymorphs be the “same” as the reference listed drug (RLD) in active ingredients, bioavailability, and bioequivalence.
The guidance pertains to orally available drugs that are either solid- or suspension-dosage products.
Polymorphisms arise when compounds are identical chemically, but not structurally. This can happen when two solids take on different crystalline forms—such as graphite and diamond; when molecules are disordered and fail to produce a repeatable crystal lattice, as is the case for the molecules in glass; or when solvent is trapped inside the crystal structure—as in hydrates, where water molecules are found within crystals.
The guidance notes that different polymorphisms may alter physical properties of compounds and affect their solubility, which in turn can alter their bioavailability or bioequivalence. In addition, polymorphic forms of a compound may alter the way the compound behaves during production, which again, may alter the finished drug’s biological activities.
On this latter point, the guidance specifically states, “Since an ANDA applicant should demonstrate that the generic drug product can be manufactured reliably using a validated process, we recommend that you pay close attention to polymorphism as it relates to pharmaceutical processing.”
The guidance also emphasizes the effect polymorphisms may have on drug stability, which again, may alter the drug’s biological activity. But the guidance goes on to say that “it is the stability of the drug product and not stability of the drug substance polymorphic form that should be the most relevant measure of drug equality.” Otherwise, a generic drug can be considered the “same” as the active ingredient in an RLD if the generic compound conforms to the standards set out in a United States Pharmacopeia (USP) monograph, if one exists for that particular drug substance.
These standards generally include the chemical name, empirical formula, and molecular structure of the compound. However, the “FDA may prescribe additional standards that are material to the sameness of a drug substance.” But as concerns polymorphisms, the guidance goes on to say “…differences in drug substance polymorphic forms do not render drug substances different active ingredients for the purposes of ANDA approvals….”
Finally, the guidance reminds ANDA applicants that the biological performance characteristics of a drug are also dependent on the drug’s formulation and advises applicants to consider the properties of both the drug substance and formulation excipients, when assessing “sameness.”
A sponsor of an Abbreviated New Drug Application (ANDA) must have information to show that the proposed generic product and the innovator product are both pharmaceutically equivalent and bioequivalent, and therefore, therapeutically equivalent.
Many pharmaceutical solids exist in several crystalline forms and thus exhibit polymorphism. Polymorphism may result in differences in the physico-chemical properties of the active ingredient and variations in these properties may render a generic drug product to be bioinequivalent to the innovator brand. For this reason, in ANDAs, careful attention is paid to the effect of polymorphism in the context of generic drug product equivalency.
This review ..Adv Drug Deliv Rev. 2004 Feb 23;56(3):397-414……discusses the impact of polymorphism on drug product manufacturability, quality, and performance. Conclusions from this analysis demonstrate that pharmaceutical solid polymorphism has no relevance to the determination of drug substance “sameness” in ANDAs.
Three decision trees for solid oral dosage forms or liquid suspensions are provided for evaluating when and how polymorphs of drug substances should be monitored and controlled in ANDA submissions. Case studies from ANDAs are provided which demonstrate the irrelevance of polymorphism to the determination of drug substance “sameness”. These case studies also illustrate the conceptual framework from these decision trees and illustrate how their general principles are sufficient to assure both the quality and the therapeutic equivalence of marketed generic drug products.
read
ANDAs: Pharmaceutical Solid Polymorphism – Food and Drug … click here
also
Issues of Polymorphism and Abbreviated New Drug Applications click here
and
POLYMORPHISM OF DRUGS – Seventh Street Development Group click here
An Overview of Solid Form Screening During Drug … – ICDD..http://www.icdd.com/ppxrd/10/presentations/PPXRD-10_Ann_Newman.pdf
http://www.ivtnetwork.com/sites/default/files/Polymorphism_01.pdf
Although polymorph/salt screening should ideally be performed to select the optimum solid form upon selection of the lead compound prior to animal pharmacokinetic (PK) studies, these screening study can be costly and time consuming. But the consequences of late discovery of a thermodynamic form are grave, so there must be a strategy to minimize the risk without spending a large amount of resources.
We find this right strategy based on early BCS classification of new compounds. We tailor the upfront polymorph/salt studies based on the risk in bioavailability, stability and manufacture-ability. Since regulatory agencies worldwide require the use of the same salt across preclinical and clinical studies, for insoluble or unstable compounds, salt screening is done early to enable further compound development.
Once salt is selected, the polymorph screening of the selected salt if soluble may be done a little later after animal study. However it is paramount to confirm 1) the polymorph in use is stable in the toxicological vehicle, 2) no changes of solid forms during shipping and storage, 3) no significant degradation upon storage.
Should there be polymorphic changes such as formation of a hydrate in the animal vehicle resulting in lowered solubility and precipitation of the hydrate, or formation of a hydrate when exposed to humidity during shipping and storage, early discovery of the stable forms will enable consistent animal exposure and avoid study repeats and delays in timelines.
Therefore, although most companies do not perform comprehensive polymorph screening until late in the development cycle, we recommend identification of a thermodynamic stable form within the confine of not only the API manufacture processes but also in the designated animal and human formulations.
For instance, for a drug product manufactured by direct compression, the solidstate properties of the active ingredient will likely be critical to the manufacture of the drug product, particularly when it constitutes the bulk of the tablet mass.
On the other hand, for a drug product manufactured by wet granulation, the solidstate properties of the active ingredient may no longer be important but the potential for polymorphic conversion is high in the presence of high moisture contents. In the context of the effect of polymorphism on pharmaceutical processing, what is most relevant is the ability to consistently manufacture a drug product that conforms to applicable in-process controls and release specifications.
This upfront work is especially critical to insoluble compounds prone to varied oral bioavailability in animal and human.
ASPARAGUS AND THE SMELL
 ASPARAGUS
ASPARAGUS

Asparagusic acid

Asparagusic acid is the organosulfur with the formula S2(CH2)2CHCO2H. The molecule contains both carboxylic acid and disulfide functional groups. It is present in the vegetable asparagus and may be the metabolic precursor to other odorous thiol compounds.
The material was originally isolated from an aqueous extract of asparagus.

Biosynthetic studies revealed that asparagusic acid is derived from isobutyric acid. This colorless solid has a melting point (m.p.) of 75.7–76.5 °C. The corresponding dithiol (m.p. 59.5–60.5 °C) is also known; it is called dihydroasparagusic acid or dimercaptoisobutyric acid.
 3D MODEL
3D MODEL
Over the past forty years several papers have been published on the subject, and several studies undertaken, to try and determine the chemical compounds responsible, and though there is still no definitive verdict as to the manner in which these compounds are formed, it has been suggested that they all form from asparagusic acid.

Asparagusic acid is, unsurprisingly considering the name, a chemical found exclusively in asparagus, and absent in other related vegetables.
The asparagus-pee molecules that you smell come mostly from the breakdown of a molecule known as asparagusic acid, which is present naturally in asparagus. When your body breaks down asparagusic acid it forms a wide variety of chemicals, all of which contain sulfur!

This has made it an obvious candidate for being the origin of the peculiar effect that asparagus has on urine. It has been suggested by recent studies that it could be metabolised in the body to produce the volatile compounds found in the urine after consuming the vegetable.

Steamed asparagus prepared with roasted pine nuts
Many chemicals that contain sulfur atoms smell horrible in similar ways, and I have no idea why this is. This is one chemical/biological mystery that, much to my chagrin, remains unsolved in my head (internet people, if the reason is known, please help!).
Aside from sulfur, the thing that all these smelly asparagus-pee chemicals have in common is that they are “light” enough (a.k.a. they are “volatile”, which means they have a relatively low boiling point) that they can float up into the air and into your nose. That is partly why asparagus doesn’t smell like asparagus-pee, because asparagusic acid is not volatile (remember that word). In fact, asparagusic acid boils above 300 °C (>600 °F), so there is no way any of it gets into your nose!
Asparagus has been used as a vegetable and medicine, owing to its delicate flavour, diuretic properties, and more. It is pictured as an offering on an Egyptian frieze dating to 3000 BC. Still in ancient times, it was known in Syria and in Spain. Greeks and Romans ate it fresh when in season and dried the vegetable for use in winter; Romans would even freeze it high in the Alps, for the Feast of Epicurus. Emperor Augustus tossed off the “Asparagus Fleet” for hauling the vegetable, and coined the expression “faster than cooking asparagus” for quick action. A recipefor cooking asparagus is in the oldest surviving book of recipes, Apicius’s third-century AD De re coquinaria, Book III.
The ancient Greek physician Galen (prominent among the Romans) mentioned asparagus as a beneficial herb during the second century AD, but after the Roman empire ended, asparagus drew little medieval attention. until al-Nafzawi‘s The Perfumed Garden. That piece of writing celebrates its (scientifically unconfirmed) aphrodisiacal power, a supposed virtue that the IndianAnanga Ranga attributes to “special phosphorus elements” that also counteract fatigue. By 1469, asparagus was cultivated in French monasteries. Asparagus appears to have been hardly noticed in England until 1538, and in Germany until 1542.
The finest texture and the strongest and yet most delicate taste is in the tips. The points d’amour (“love tips”) were served as a delicacy to Madame de Pompadour. Asparagus became available to the New World around 1850, in the United States.

German botanical illustration of asparagus
Chemistry

Asparagus foliage turns bright yellow in autumn
Certain compounds in asparagus are metabolized to yield ammonia and various sulfur-containing degradation products, including various thiols andthioesters, which give urine a characteristic smell.
Some of the volatile organic compounds responsible for the smell are:
- methanethiol
- dimethyl sulfide
- dimethyl disulfide
- bis(methylthio)methane
- dimethyl sulfoxide
- dimethyl sulfone
Subjectively, the first two are the most pungent, while the last two (sulfur-oxidized) give a sweet aroma. A mixture of these compounds form a “reconstituted asparagus urine” odor. This was first investigated in 1891 by Marceli Nencki, who attributed the smell to methanethiol. These compounds originate in the asparagus as asparagusic acid and its derivatives, as these are the only sulfur-containing compounds unique to asparagus. As these are more present in young asparagus, this accords with the observation that the smell is more pronounced after eating young asparagus. The biological mechanism for the production of these compounds is less clear.
The onset of the asparagus urine smell is remarkably rapid. The smell has been reported to be detectable 15 to 30 minutes after ingestion.
Gas chromatography-mass spectrometry was used to analyse the ‘headspace’ of urine produced after consumption of asparagus. The headspace is the gas space immediately above the liquid surface, which is occupied by light, volatile compounds in the liquid, and analysis of this is useful in identifying odour-causing compounds. The analysis of the post-asparagus urine showed the presence of several compounds that were not present, or present in negligible amounts, in normal urine. The primary compounds present, in quantities a thousand times greater than in normal urine, were methanethiol and dimethyl sulfide. The compounds dimethyl sulfide and dimethyl sulfone were also present and it was suggested that they modify the aroma to give it a ‘sweet’ edge.
| Nutritional value per 100 g (3.5 oz) | |
|---|---|
| Energy | 85 kJ (20 kcal) |
| Carbohydrates | 3.88 g |
| – Sugars | 1.88 g |
| – Dietary fibre | 2.1 g |
| Fat | 0.12 g |
| Protein | 2.2 g |
| Vitamin A equiv. | 38 μg (5%) |
| – beta-carotene | 449 μg (4%) |
| – lutein and zeaxanthin | 710 μg |
| Thiamine (vit. B1) | 0.143 mg (12%) |
| Riboflavin (vit. B2) | 0.141 mg (12%) |
| Niacin (vit. B3) | 0.978 mg (7%) |
| Pantothenic acid (B5) | 0.274 mg (5%) |
| Vitamin B6 | 0.091 mg (7%) |
| Folate (vit. B9) | 52 μg (13%) |
| Choline | 16 mg (3%) |
| Vitamin C | 5.6 mg (7%) |
| Vitamin E | 1.1 mg (7%) |
| Vitamin K | 41.6 μg (40%) |
| Calcium | 24 mg (2%) |
| Iron | 2.14 mg (16%) |
| Magnesium | 14 mg (4%) |
| Manganese | 0.158 mg (8%) |
| Phosphorus | 52 mg (7%) |
| Potassium | 202 mg (4%) |
| Sodium | 2 mg (0%) |
| Zinc | 0.54 mg (6%) |
|
Link to USDA Database entry
|
|

Garden Cress Extract Kills 97% of Breast Cancer Cells in Vitro

Garden Cress Extract Kills 97% of Breast Cancer Cells in Vitro: Garden cress, like broccoli, is a cruciferous-family vegetable but is unique because it contains very high amounts of BITC (benzyl isothiocyanate) which has emerged as a powerful anti-cancer compound. In this study, BITC was seen to kill 97% of ER- breastcancer cells (MDA-MB-231) after 24 hours of treatment. For comparison, the same dose of sulforaphane from broccoli killed only 75% of the cancer cells.

In other research, BITC has been found to slow the rate of breast cancer metastasizing by 86% and when given to mice, resulted in breast tumors 53% smaller than in untreated mice. BITC is now being intensively studied for a variety of cancers and has been shown in lab studies to be active against melanoma, glioma, prostate cancer, lung cancer, ovarian cancer, pancreatic cancer and others. Garden cress is one of the best sources of BITC. Other good sources include cabbage, Indian cress, Japanese radish (in particular Karami daikon) and, quite surprisingly, papaya seeds. As with othercruciferous vegetables, the best way to eat cress is raw in order to maximize the delivery of BITC.
http://www.ncbi.nlm.nih.gov/pubmed/17121941
http://extension.usu.edu/files/publications/publication/HG_Garden_2006-05.pdf
and
http://nopr.niscair.res.in/bitstream/123456789/12732/1/IJNPR%202(3)%20292-297.pdf
BITC

Botanical name: Lepidium sativum L.
Family: Brassicaceae = Cruciferae
Common names. English: cress, common cress, garden cress, land cress, pepper cress; Spanish: mastuerzo, mastuerzo hortense, lepidio, berro de jardín (Spain), berro de sierra, berro hortense (Argentina), escobilla (Costa Rica); Catalan: morritort, morrisà, Portuguese and Galician: masturco, mastruco, agrião-mouro, herba do esforzo; Portuguese: mastruco do Sul, agrião (Brazil); Basque: buminka, beatzecrexu
Synonyms/Common Names/Related Substances:
- Alpha-linolenic acid (ALA), agrião (Portuguese), agrião-mouro (Portuguese, Galician), beatzecrexu (Basque), berro de jardín (Spanish), berro de tierra (Spanish), berro hortense (Spanish), benzyl isothiocyanate (BITC), Brassicaceae (family), bran, buminka (Basque), common cress, cress, docosahexaenoic acid (DHA), eicosapentaenoic acid (EPA), escobilla (Spanish), endosperm, fiber, garden cress seed oil (GCO), garden pepper grass, glucosinolates, glutamic acid, herba do esforzo (Portuguese, Galician), hurf (Arabic), indoles, isothiocyanates, kardamon (Greek), land cress, linoleic acid (LA), lectin, lepidio (Spanish), Lepidium sativium, Lepidium sativum, leucine, mastruco (Portuguese, Galician), mastruco do sul (Portuguese), mastuerzo (Spanish), mastuerzo hortense (Spanish), methanol, morrisá (Catalan), morritort (Catalan), nasturtium (Latin), nasum torcere (Latin), omega-3 fatty acid, pepper cress, pepper grass, pepperwort, sulforaphane, tuffa’ (Arabic), turehtezuk (Persian), water cress, whole meal.
- Combination product example: SulforaWhite (a liposomal preparation that contains Lepidium sativum sprout extract, glycerin, lecithin, phenoxyethanol, and water).
Garden cress [commonly known as aliv in Marathi or halim in Hindi] is a green, cool-season perennial plant used as a leafy vegetable, typically used as a garnish. Undisturbed, the plant can grow to a height of two feet with minimal maintenance. When mature, garden cress produces white or light-pink flowers, and small seed pods. It has long leaves at the bottom of the stem and small, bright-green, feather-like leaves arranged on opposite sides of its stalks at the top.
Garden Cress, also called Pepper Wort, is an herb that is botanically known as Lepidium Sativum. It is referred to as ‘Aliv’ in Marathi and ‘Halim’ in Hindi. Belonging to the family Cruciferae, it is grown in all parts of India and is often used in the Indian cuisine. The leaves, roots, as well as seeds of this plant are used in cooking as they are extremely nutritious and also therapeutic in nature. The flowers of this plant are either white or light-pink in color.
This herb is the best source of iron and is hence recommended in the treatment of iron-deficiency anemia. It is also rich in folate, calcium, ascorbic acid, tocopherol, and beta-carotene. Garden Cress seeds are loaded with not just protein, but also linoleic and arachidic fatty acids. Since they contain phytochemicals that mimic estrogen to some extent, intake of these seeds is known to regulate menstruation and stimulate milk production in lactating mothers. That is precisely why women are given foods containing Garden Cress following childbirth.
The blood-purifying as well as antioxidant properties of this amazing plant are well documented. Hence, its regular consumption can greatly help to boost one’s immunity and prevent a gamut of diseases. It acts as a general tonic and can also help to increase the libido naturally. Since the testae of these seeds contain mucilage, they are invaluable in the management of both dysentery and constipation. The whole plant, along with its seeds, is said to be good for the eyes too. Hence, it is advisable to add it raw to salads, sandwiches, and chutneys, or to simply use it as a garnish along with coriander leaves for any food item.
Pregnant women should avoid taking Garden Cress in any form because it has the ability to induce uterine contractions and thereby trigger a spontaneous abortion. Also, since it is goitrogenic in nature, it may not be suitable for patients suffering from hypothyroidism. The oil derived from Garden Cress seeds is edible and can therefore be used as a cooking medium; however, some people may experience symptoms of indigestion due to its use. Such individuals should discontinue using this oil or mix it with some other edible oil, so as to dilute it and reduce its adverse effects.
Cress (Lepidium sativum), sometimes referred to as garden cress to distinguish it from similar plants also referred to as cress, is a rather fast-growing, edible herb. Garden cress is genetically related to watercress and mustard, sharing their peppery, tangy flavor and aroma. In some regions, garden cress is known as mustard and cress, garden pepper cress, pepperwort pepper grass, or poor man’s pepper.[1][2]
This annual plant can reach a height of 60 cm (~24 inches), with many branches on the upper part. The white to pinkish flowers are only 2 mm (1/12 of an inch) across, clustered in branched racemes.[3][4]
Origin of the name
Cultivation of this species, which is native to Southwest Asia (perhaps Persia) and which spread many centuries ago to western Europe, is very old, as is shown by the philological trace of its names in different Indo-European languages. These include the Persian word turehtezuk, the Greek kardamon, the Latin nasturtium and Arabic tuffa’ and hurf. In some languages there is a degree of confusion with watercress. It seems that the meaning of the word nasturtium (nasum torcere, because its smell causes the nose to turn up) must have been applied initially to garden cress, as both Pliny and Isidoro de Sevilla explain. The confusion remains with the terms used by the Hispano-Arabs. The word hurf is applied without distinction to watercress and garden cress (several species certainly of up to three different genera: Nasturtium, Lepidium and Cardaria). Thus the medieval agronomists of Andalusia went as far as differentiating between several hurf, such as hurf abyad, hurf babili, hurf madani….
Garden cress in agriculture
Garden cress is commercially grown in England, France, the Netherlands and Scandinavia.[5]
Cultivation of garden cress is practical on both mass scales and on the individual scale. Garden cress is suitable for hydroponic cultivation and thrives in slightly alkaline water. In many local markets, the demand for hydroponically grown cress can exceed available supply, partially because cress leaves are not suitable for distribution in dried form, so can be only partially preserved. Consumers commonly acquire cress as seeds or (in Europe) from markets as boxes of young live shoots.[5]
Edible shoots are typically harvested in one to two weeks after planting, when they are 5–13 cm (2 – 5 inches) tall.[6]
Properties, uses and cultivation
Xenophon (400 BC) mentions that the Persians used to eat this plant even before bread was known. It was also familiar to the Egyptians and was very much appreciated by the Greeks and Romans, who were very fond of banquets rich in spices and spicy salads. Columela (first century) makes direct reference to the cultivation of garden cress. In Los doce libros de Agricultura, he writes: ” …immediately after the calends of January, garden cress is sown out… when you have transplanted it before the calends of March, you will be able to harvest it like chives, but less often… it must not be cut after the calends of November because it dies from frosts, but can resist for two years if it is hoed and manured carefully… there are also many sites where it lives for up to ten years” (Book XI). The latter statements seem to indicate that he is also speaking of the perennial species L. Iatifolium, as L. sativum is an annual.
Almost all of the Andalusian agronomists of the Middle Ages (Ibn Hayyay, Ibn Wafid, Ibn al-Baytar, Ibn Luyun, Ibn al-Awwam) and many of the doctors, such as Maimonides, mention garden cress. Ibn al-Awwam also includes references from Abu al-Jair, Abu Abdalah as well as from Nabataean agriculture and, among other comments, he says: “Garden cress is sown between February and April (in January in Seville). It has small seeds which are mixed with earth for sowing to prevent the wind carrying them away…. It is harvested in May and is grown between ridges, in combination/conjunction with flax cultivation.”
Many of the authors of the old oriental and Mediterranean cultures emphasized the medicinal properties of cress, especially as an antiscorbutic, depurative and stimulant. Columela notes its vermifugal powers. Ibn al-Awwam refers to certain apparently antihistaminic properties, since it was used against insect bites and also as an insect repellent, in the form of a fumigant. It was perhaps Ibn al-Baytar, an Andalusian botanist (eighth century), who collected most information on its properties, summarizing the opinions of other authors such as El Farcy, who says that it incites coitus and stimulates the appetite; Ibn Massa, according to whom it dissipates colic and gets rid of tapeworms and other intestinal worms; or Ibn Massouih, who mentions that it eliminates viscous humours. Ibn al-Baytar also says that it is administered against leprosy, is useful for renal “cooling” and that, if hair is washed with garden cress water, it is “purified” and any loss is arrested.
In Iran and Morocco, the seeds are used as an aphrodisiac. In former Abyssinia, an edible oil was obtained from the seeds. In Eritrea, it was used as a dyestuff plant. Some Arab scholars have attributed garden cress’s reputation among Muslims to the fact that it was directly recommended by the Prophet.
Garden cress’s main use was always as an aromatic and slightly pungent plant. Not only in antiquity but also in the Middle Ages it enjoyed considerable prestige on royal tables. The young leaves were used for salads. The ancient Spartans ate them with bread. This use still continues and they are also eaten with bread and butter or with bread to which lemon, vinegar or sugar is added. However, it is mainly used nowadays in the seedling stage, the succulent hypocotyls being added to salads and as a garnish and decoration for dishes.
The roots, seeds and leaves have been used as a spicy condiment. Columela explains how oxygala, a type of curd cheese with herbs, was prepared: “Some people, after collecting cultivated or even wild garden cress, dry it in the shade and then, after removing the stem, add its leaves to brine, squeezing them and placing them in milk without any other seasoning, and adding the amount of salt they consider sufficient…. Others mix fresh leaves of cultivated cress with sweetened milk in a pot…”.
L. Iatifolium L. stands out for its horticultural interest; although it grows spontaneously on the edges of rivers and lakes, it is also occasionally grown in the same way as L. sativum. Its young leaves can be used for salads; the ancient Greeks and Romans used to grow it for this purpose. Its leaves and seeds were also used as a spicy condiment. Several sauces are prepared with its leaves, including in particular the bitter sauce of the paschal lamb of the Jews. The seeds of this species were known in England as the poor people’s pepper. The roots have been used on occasion as a substitute for radish.
In the fifteenth century, we know through Alonso de Herrera that garden cress was one of the vegetables most widely eaten in Castile. During the sixteenth century, obstinate attempts were made to introduce it into America. Right up to the beginning of the nineteenth century, its cultivation in Spain continued to be important, since Boutelou and Boutelou (1801) deal specifically with this crop in their Tratado de la huerta, commenting on the existence of several cultivars. At present, the cultivation of cress is very occasional in countries such as Spain and France. Water cress, in competition with garden cress, has eclipsed the cultivation of the latter. However, this is not the case in other central European countries or the United Kingdom, where its use is normal and the system of cultivation has changed substantially.
Botanical description
Cress is an annual, erect herbaceous plant, growing up to 50 cm. The basal leaves have long petioles and are lyrate-pinnatipartite; the caulinar leaves are laciniate-pinnate while the upper leaves are entire. The inflorescences are in dense racemes. The flowers have white or slightly pink petals, measuring 2 mm. The siliquae measure 5 to 6 x 4 mm, are elliptical, elate from the upper half, and glabrous. Cress flowers in the wild state between March and June.
It is an allogamous plant with self-compatible and self-incompatible forms and with various degrees of tolerance to prolonged autogamy. There are diploid forms, 2n = 2x = 16, and tetraploid forms, 2n = 4x =32. A degree of variability is noted in the character of the basal leaves which are cleft or split to a greater or lesser degree, a character which is controlled by a single incompletely dominant gene.
Ecology and phytogeography
Cress is a plant that is well suited to all soils and climates, although it does not tolerate frosts. In temperate conditions, it has a very rapid growth rate. It grows subspontaneously in areas transformed by humans, close to crops or human settlements. It appears in this way on the Iberian peninsula, mainly in the eastern regions.
Wild cress extends from the Sudan to the Himalayas. Most authors consider it to be a native of western Asia, whence it passed very quickly to Europe and the rest of Asia as a secondary crop, probably associated with cultivars of flax. Vavilov considers its main centre to be Ethiopia, where he found the widest variability; the Near East, central Asia and the Mediterranean are considered secondary centres. It is now naturalized in numerous parts of Europe, including the British Isles.
Cress in cookery
Genetic diversity
The genus Lepidium is made up of about 150 species, distributed throughout almost all temperate and subtropical regions of the world. On the Iberian peninsula and the Balearic Islands, at least 20 species or subspecies exist among the autochthonous and allochthonous taxa, some genetically close to L. sativum. Seven of them are exclusively endemic to the peninsula or, at the very most, are common with North Africa. Other close species are L. campestre (L.) R. Br. and L. ruderale L. which also have edible leaves. The leaves of L. campestre are used to prepare excellent sauces for fish.
Common cress (L. sativum L.), with regard to the anatomy of the leaf, stem and root, has been divided into three botanical varieties: vulgare, crispum and latifolium. The latter is the most mesomorphic, crispum the most xeromorphic and vulgare intermediate.
At present, most of the studies on the variability and development of new cultivars are being carried out in liaison with the VIR of St Petersburg, where there is a good collection of material. Of the 350 forms of garden cress studied in the Ukraine, Uzkolistnyti 3 was the best, being highly productive and of good quality. It is being used as the basis of improvement programmes, as it appreciably surpasses the best Soviet varieties in production and quality. Other cultivars well suited to European Russia are Tuikers Grootbladige (broad-leaved) and the lines Mestnyi k 137, k 106 and k 115. Of the types most cultivated in Europe, Early European, Eastern, Dagestan and Entire Leaved stand out, being distinguished by the length and shape of the leaf, earliness and susceptibility to cold. In Western Europe, one broad leaved type is especially appreciated (Broad Leaved French) as are curly types (Curly Leaved), the latter being used extensively to garnish dishes. In Africa, there are red, white and black varieties.
This crop is also arousing interest in Japan, and collecting expeditions to Nepal have been organized. Some specimens collected during an expedition to Iraq in 1986 are now stored in Abu Ghraib and in Gratersleben, Germany. There are also small collections of L. sativum in the PGRC in Addis Ababa (Ethiopia), at the ARARI of Izmir in Turkey and in Bari, Italy. At the Universidad Politécnica de Madrid there are accessions of 20 species of Lepidium, while the BGV of the Córdoba Botanical Garden keeps germplasm of the southern Iberian species of the genus.
| Nutritional value per 100 g (3.5 oz) | |
|---|---|
| Energy | 134 kJ (32 kcal) |
| Carbohydrates | 5.5 g |
| – Sugars | 4.4 g |
| – Dietary fiber | 1.1 g |
| Protein | 2.6 g |
| Vitamin A equiv. | 346 μg (43%) |
| – beta-carotene | 4150 μg (38%) |
| – lutein and zeaxanthin | 12500 μg |
| Thiamine (vit. B1) | 0.08 mg (7%) |
| Riboflavin (vit. B2) | 0.26 mg (22%) |
| Niacin (vit. B3) | 1 mg (7%) |
| Pantothenic acid (B5) | 0.247 mg (5%) |
| Vitamin B6 | 0.247 mg (19%) |
| Folate (vit. B9) | 80 μg (20%) |
| Vitamin C | 69 mg (83%) |
| Vitamin E | 0.7 mg (5%) |
| Vitamin K | 541.9 μg (516%) |
| Calcium | 81 mg (8%) |
| Iron | 1.3 mg (10%) |
| Magnesium | 38 mg (11%) |
| Manganese | 0.553 mg (26%) |
| Phosphorus | 76 mg (11%) |
| Potassium | 606 mg (13%) |
| Link to USDA Database entry Percentages are roughly approximated using US recommendations for adults. Source: USDA Nutrient Database |
|
Garden cress is added to soups, sandwiches and salads for its tangy flavor.[6] It is also eaten as sprouts, and the fresh or dried seed pods can be used as a peppery seasoning (haloon).[5] In England, cut cress shoots are commonly used in sandwiches with boiled eggs, mayonnaise and salt.

Garden cress can grow almost anywhere.
Nutrition profile
Garden cress is an important source of iron, folic acid, calcium, vitamins C, E and A. The seed contains arachidic and linoleic fatty acids. The seeds are high in calories and protein, whereas the leaves are an excellent source of vitamin A, C and folate.
| Energy | 30 Kcal |
| Carbohydrates | 5.5 g |
| Dietary fibre | 1.1 g |
| Protein | 2.6 g |
| Fat | 0.7 g |
| Vitamin A | 346 mcg |
| Folate | 80 mcg |
| Vitamin C | 69 mg |
| Calcium | 81 mg |
| Iron | 1.3 mg |

Both the leaves and stems of cress can be eaten raw in salads or sandwiches, and are sometimes called cress sprouts. When buying cress, look for firm, evenly coloured, rich green leaves. Avoid cress with any signs of slime, wilting, or discoloration. If stored in plastic, it can last up to five days in a refrigerator. To prolong the life of cress, place the stems in a glass container with water and cover them, refrigerating the cress until it is needed.
Cultivation practices
Cress is an easily grown plant with few requirements. It can be broadcast after the winter frosts or throughout the year in temperate climates. However, Boutelou and Boutelou (1801) were already recommending sowing in shallow furrows, which enables surplus plants to be thinned out and facilitates hoeing. Sowing has to be repeated every 15 to 20 days so that there is no shortage of young shoots and new leaves for salads – the leaves of earlier sowings begin to get tough and are no longer usable. The seed sprouts four or six days after sowing, depending on the season, and the leaves are ready for consumption after two or three weeks.
The usual form of cultivation continues to be as described, with 15 to 20 cm between rows and the use of irrigation in the summer, since they are lightly rooted seedlings which can dry up in a few days. Its growth is very rapid and harvesting can begin in the same month as sowing, with yields reaching 6 tonnes per hectare.
Health benefits of garden cress
For women’s health
Emenagogue: Garden cress has mild oestrogenic properties. It helps to regulate the menstrual cycle.
Galactogogue: Kheer made of garden cress seeds increases milk production and secretion in lactating mothers. Because of its high iron and protein content, it is often given post-partum to lactating mothers.
Aphrodisiac: Garden cress helps to improve libido.
For the gastro intestinal tract
Garden cress helps purify blood and stimulate appetite. It is used during constipation as a laxative and a purgative. Paste made of the seeds can be taken internally with honey to treat amoebic dysentery. The mucilage of the germinating seeds allays the irritation of the intestines in dysentery and diarrhoea. Garden cress crushed and drunk with hot water is beneficial to treat colic especially in infants.
For the respiratory tract
Garden cress seeds are good expectorants and when chewed they treat sore throat, cough, asthma and headache. The aerial parts are used in the treatment of asthma and cough.
For anaemia
Garden cress seeds being the richest source of non-haeme iron [iron found in haemoglobin which is an easily absorbed dietary iron.] help to increase the haemoglobin levels. When taken regularly, it helps to alleviate anaemia. It is advisable to have vitamin C half an hour after consumption of these seeds as it enhances iron absorption.

For diabetes
The seed coat of germinating seeds contains mucilage, which has a phytochemical called lepidimoide. Studies show that seeds of the plant lower the glycemic response to a test meal.
note sodium 2-O-rhamnopyranosyl-4-deoxy-threo-hex-4-enopyranosiduronate (designated lepidimoide)
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1075667/
epi-Lepidimoide
Sodium 6-deoxy-2-O-(4-deoxy-β-L-threo-hex-4-enopyranuronosyl)-α-L-glucopyranose
 cas 145039-76-5 and 157676-09-0
cas 145039-76-5 and 157676-09-0
The total synthesis of the unsaturated disaccharide, lepidimoide 4-deoxy--l-threo-hex-4-enopyranuronosyl-(1->2)-l-rhamnopyranose sodium salt, has been carried out from d-glucose and l-rhamnose (Tetrahedron Lett. 1993, 34, 2653), but the process is very long and complicated. A method for more easily producing this compound and in large quantities is necessary for further research. We have succeeded in conveniently synthesizing lepidimoide from okra (Hibiscus esculentus L.) fruit mucilage. At the same time, the isomer (epi-lepidimoide) was obtained as a byproduct. The structure was determined as the 4-deoxy--l-threo-hex-4-enopyranuronosyl-(1->2)-6-deoxy-l-glucopyranose sodium salt by spectral analysis. We found that lepidimoide easily epimerized to epi-lepidimoide in alkaline media. Both lepidimoide and epi-lepidimoide exhibited the same high activity in the cockscomb hypocotyls elongation test….Carbohydrate Research, Volume 339, Number 1, 2 January 2004 , pp. 9-19(11)
-
Dictionary of Natural Products, Supplement 3
books.google.co.in/books?isbn=0412604302John Buckingham – 1996 – ScienceLepidimoide. L-30020. 6-Deoxy-2-0-(4-deoxy-fi-L-lhreo-hex-4-enopyranuronosyl)-L- mannose, 9CI [157676-09-0] HOA—O …
lepidimoide
Sodium 2-O-L-rhamnopyranosyl-4-deoxy-alpha-L-threo-hex-4-eno-pyranosiduronateMolecular Formula: C12H17NaO10 Molecular Weight: 344.247149sodium;(2S,3R,4S)-3,4-dihydroxy-2-[(2S,3R,4R,5R,6S)-2,4,5-trihydroxy-6-methyloxan-3-yl]oxy-3,4-dihydro-2H-pyran-6-carboxylateSodium2-O-L-rhamnopyranosyl-4-deoxy-α-L-threo-hex-4-eno-pyranosiduronate
For cancer
Garden cress seeds contain antioxidants like vitamin A and E which help protect cells from damage by free radicals. Hence, these seeds have a chemoprotective [drugs which protect healthy tissue from the toxic effects of anticancer drugs] nature.
Anti-Cancer:
Being a family of Brassica family it has good anti cancer property. Garden cress seeds contain antioxidants like vitamin A and E which help protect cells from damage by free radicals. Hence, these seeds have a chemo protective nature.

Few years back garden cress seeds/ halim/ aliv was not a common food item or a familiar to be heard. But as years passed it’s popularity and it’s importance have been realized and now people are aware of some of the facts of these seeds. Though these facts are also accompanied by some myths. So I chose to write and clear few myths and doubts of these seeds so that maximum people can make use of it in their lives and improve quality of their diet and nutrition.
Nutritive value of these seeds is very high. It is available in almost all parts of the world. Its high nutritive value and cheaper availability makes it possible for people of all the sections of society to include in the diet and increase nutritive value of their meals. Garden cress seeds are very high in Iron and Folic acid content. These seeds are use as herbal medicine to treat iron deficiency anemia. People consuming 2tsp/day have seen to have good increased levels of hemoglobin over a period of 1-2 months. Garden cress seeds also contains calcium, ascorbic acid, tocopherol, and beta-carotene which helps to improve body’s immunity.Garden Cress seeds are loaded with not just protein, but also linoleic and arachidic fatty acids. Since they contain phytochemicals that resemble estrogen to some extent, intake of these seeds helps to regulate menstruation and stimulate milk production in lactating mothers. That is why women are given foods containing Garden Cress following childbirth.
Traditionally garden cress seeds were considered to be useful only during last few weeks of gestation and post delivery. It is considered to be hot food. But the truth is that these seeds have ability too increase uterine contraction. So in later stages of pregnancy it helps in inducing labour but if in case consumed in early stage of pregnancy (1st trimester) it leads to spontaneous abortion. It is also very carefully prescribed to a hypothyroid patients because it belongs to cruciferous family and is a goitrogen that prevent iodine absorption.

How to eat:
1. Roasted slightly with added salt.
2. Soaked in water then added to milk or juice.
3. Chikki or laddoo can be made. (preparation similar to til laddoo/chikki).
How much to eat:
Start with 1 tsp/day and then an be taken 1 tsp/2c a day.
Cress seeds have many more medicinal properties and researches are still on to find its benefits on health. Garden cress should be eaten in moderation. Excess consumption of these seeds may hv adverse effect on health.
For other things
Garden cress seeds are memory boosters because they contain arachidic and linoleic acids. They help gaining lean body mass because they are a good source of iron and protein. Research has proved that 60 per cent women have hair loss due to low iron levels and poor protein. A teaspoon of garden cress seeds soaked in lime water helps in iron absorption, which in turn strengthens hair. The plant is also used in treating bleeding piles. The leaves are mildly stimulant and diuretic, useful in scorbutic [related to or resembling scurvy] diseases and liver complaints. A paste of the seeds with water is applied to chapped lips, and against sunburn.
Side-effects
It is an abortifacient [substance that induces abortion], if had in excess. It contains goitrogens that prevent iodine absorption in thyroids and hence can lead to hypothyroidism. If large quantities of garden cress are consumed, the mustard oil it contains may cause digestive difficulties in some people who are sensitive to it. Therefore, garden cress should be eaten in moderation.
Other uses
Garden cress, known as chandrashoor, and the seeds, known as halloon[7] in India, are commonly used in the system of Ayurveda to prevent postnatal complications.[citation needed]
Garden cress seeds, since ancient times, have been used in local traditional medicine of India.[8] Garden cress seeds are bitter, thermogenic, depurative, rubefacient, galactogogue, tonic, aphrodisiac, ophthalmic, antiscorbutic, antihistaminic and diuretic. They are useful in the treatment ofasthma, coughs with expectoration, poultices for sprains, leprosy, skin disease, dysentery, diarrhoea, splenomegaly, dyspepsia, lumbago, leucorrhoea, scurvy and seminal weakness. Seeds have been shown to reduce the symptoms of asthma and improve lung function in asthmatics.[9]The seeds have been reported as possessing a hypoglycemic property[10] and the seed mucilage is used as a substitute for gum arabic and tragacanth.
Cress may be given to budgerigars.[11] The seeds are employed as poultice for removing pain, swelling etc.Some use it in the belief that it can cure asthma, bronchitis bleeding piles.[12]
Some use Lepidium sativum seeds for indigestion and constipation.[13]
Prospects for improvement
Most of the genetic improvement work on garden cress is being carried out in the CIS, with little or no work being done at present in the countries of western Europe. Mainly early cultivars with a prolonged production period and better cold tolerance are being developed.
Cress can be grown and used like white mustard. It germinates more slowly at low temperatures, the emergence period being three or four days longer. Shortening this period is an interesting improvement objective.
However, cress’s recovery and its greater presence on markets mainly depends on a modification of cultivation and marketing techniques. In countries such as the United Kingdom, where this vegetable is normally to be found at the markets, cultivation takes place in greenhouses throughout the year. The whole succulent hypocotyls of the very young seedlings are eaten. The seed is placed on the soil surface on soft, level beds. It is finely sprinkled with water and then covered with sackcloth which has been steam-sterilized and moistened. The latter is frequently wetted to maintain moisture and is removed when the seedlings reach 4 to 5 cm in height (after approximately seven days in spring and autumn and ten days in winter). The yellowish leaves turn green after two to three days.
The cress is harvested when the first pair of cotyledon leaves have developed and it is marketed in small bags or trays, sometimes together with seedlings of white mustard.
Garden cress and white pepper are sometimes sown in the plastic trays or bags in which they will be sold, generally in peat with a nutrient solution.
References
- Cassidy, Frederic Gomes and Hall, Joan Houston. Dictionary of American regional English, Harvard University Press, 2002. Page 97. ISBN 0-674-00884-7, ISBN 978-0-674-00884-7
- Staub, Jack E, Buchert, Ellen. 75 Exceptional Herbs for Your Garden Published by Gibbs Smith, 2008. ISBN 1-4236-0251-X, 9781423602514
- Vegetables of Canada. Published by NRC Research Press. ISBN 0-660-19503-8, ISBN 978-0-660-19503-2
- Boswell, John T. and Sowerby, James. English Botany: Or, Coloured Figures of British Plants. Robert Hardwicke, 1863. Page 215.
- Vegetables of Canada. NRC Research Press. ISBN 0-660-19503-8, ISBN 978-0-660-19503-2
- Hirsch, David P.. The Moosewood Restaurant kitchen garden: creative gardening for the adventurous cook. Ten Speed Press, 2005. ISBN 1-58008-666-7, ISBN 978-1-58008-666-0
- http://www.organicindia.com/PR_OH_chandrashoor.php
- The Wealth of Indian Raw Materials ,. New Delhi: Publication and information Directorate. 1979. pp. CSIR Vol 9, Page 71–72.
- NP, Archana; Anita, AM (2006). “A study on clinical efficacy of Lepidium sativum seeds in treatment of bronchial asthma”. Iran J Pharmacol Ther 5: 55–59.
- M, Eddouks; Maghrani M, Zeggwagh NA, Michel JB (2005). “Study of the hypoglycaemic activity of Lepidium sativum L. aqueous extract in normal and diabetic rats”. J Ethnopharmacol 97: 391–395.
- Budgerigars – Diets, PDSA.
- Bhatiya, KN (1996). Modern Approach to Batany. India: Surya publications. p. 516.
- Najeeb-Ur-Rehman, Mehmood MH, Alkharfy KM, Gilani AU, “Prokinetic and laxative activities of Lepidium sativum seed extract with species and tissue selective gut stimulatory actions. J Ethnopharmacol. 2011 Feb 2;







ERTUGLIFLOZIN

ERTUGLIFLOZIN, PFIZER
THERAPEUTIC CLAIM Treatment of type 2 diabetes
CHEMICAL NAMES
1. β-L-Idopyranose, 1,6-anhydro-1-C-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-5-C-(hydroxymethyl)-
2. (1S,2S,3S,4R,5S)-5-{4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl}-1-(hydroxymethyl)-6,8-dioxabicyclo[3.2.1]octane-2,3,4-triol
PF-04971729, MK 8835
M. Wt: 436.88
Formula: C22H25ClO7
CAS No:. 1210344-57-2
Diabetes looms as a threat to human health worldwide. As a result, considerable research efforts are devoted to identify new and efficacious anti-diabetic agents lacking the side effects associated with some of the current drugs (hypoglycemia, weight gain).Inhibition of sodium-dependent glucose cotransporter 2 (SGLT2), a transporter located in the kidney, is a mechanism that promotes glucosuria and therefore, reduction of plasma glucose concentration. Since the mechanism operates in a glucose-dependent and insulin-independent manner, and is associated with weight loss, it has emerged as a very promising approach to the pathophysiologic treatment of type 2 diabetes. Ertugliflozin (PF-04971729), an anti-diabetic agent currently in development (Phase 3 clinical trials) and belonging to a new class of SGLT2 inhibitors bearing a dioxa-bicyclo[3.2.1]octane bridged ketal motif.

SYNTHESIS
Scheme 1 outlines the general procedures one could use to provide compounds of the present invention.

Scheme 1 AIIyI 2,3,4-tιϊ-O-benzyl-D-glucopyranoside (La, where Pg1 is a benzyl group) can be prepared by procedures described by Shinya Hanashima, et al., in Bioorganic & Medicinal Chemistry, 9, 367 (2001 ); Patricia A. Gent et al. in Journal of the Chemical Society, Perkin 1, 1835 (1974); Hans Peter Wessel in the Journal of Carbohydrate Chemistry, 7, 263, (1988); or Yoko Yuasa, et al., in Organic Process Research & Development, 8, 405-407
(2004). In step 1 of Scheme 1 , the hydroxymethylene group can be introduced onto the glycoside by means of a Swern oxidation followed by treatment with formaldehyde in the presence of an alkali metal hydroxide (e.g., sodium hydroxide). This is referred to as an aldol-Cannizzaro reaction. The Swern oxidation is described by Kanji Omura and Daniel Swern in Tetrahedron, 34, 1651 (1978). Modifications of this process known to those of skill in the art may also be used. For example, other oxidants, like stabilized 2- iodoxybenzoic acid described by Ozanne, A. et al. in Organic Letters, 5, 2903 (2003), as well as other oxidants known by those skilled in the art can also be used. The aldol Cannizzaro sequence has been described by Robert Schaffer in the Journal of The American Chemical Society, 81 , 5452 (1959) and Amigues, E.J., et al., in Tetrahedron, 63,10042 (2007).
In step 2 of Scheme 1 , protecting groups (Pg2) can be added by treating intermediate (MD) with the appropriate reagents and procedures for the particular protecting group desired. For example, p-methoxybenzyl (PMB) groups may be introduced by treatment of intermediate (MD) with p-methoxybenzyl bromide or p-methoxybenzyl chloride in the presence of sodium hydride, potassium hydride, potassium te/t-butoxide in a solvent like tetrahydrofuran, 1 ,2-dimethoxyethane or Λ/,Λ/-dimethylformamide (DMF). Conditions involving para-methoxybenzyltrichloroacetimidate in presence of a catalytic amount of acid (e.g., trifluoromethanesulfonic acid, methanesulfonic acid, or camphorsulfonic acid) in a solvent such as dichloromethane, heptane or hexanes can also be used. Benzyl (Bn) groups may be introduced by treatment of intermediate (MD) with benzyl bromide or benzyl chloride in the presence of sodium hydride, potassium hydride, potassium te/t-butoxide in a solvent like tetrahydrofuran, 1 ,2-dimethoxyethane or Λ/,Λ/-dimethylformamide. Conditions involving benzylthchloroacetimidate in presence of a catalytic amount of acid (e.g., trifluoromethanesulfonic acid, methanesulfonic acid, or camphorsulfonic acid) in a solvent such as dichloromethane, heptane or hexanes can also be used. In step 3 of Scheme 1 , the allyl protection group is removed (e.g., by treatment with palladium chloride in methanol; cosolvent like dichloromethane may also be used; other conditions known by those skilled in the art could also be used, see T. W. Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1991 ) to form the lactol (Ld).
In step 4 of Scheme 1 , oxidation of the unprotected hydroxyl group to an oxo group (e.g., Swern oxidation) then forms the lactone (l-e).
In step 5 of Scheme 1 , the lactone (Le) is reacted with Λ/,O-dimethyl hydroxylamine hydrochloride to form the corresponding Weinreb amide which may exist in equilibrium in a closed/opened form, (l-f/l-g). The “Weinreb amide” (LgJ can be made using procedures well known to those of skill in the art. See, Nahm, S., and S. M. Weinreb, Tetrahedron Letters. 22 (39), 3815-1818 (1981 ). For example, intermediate (l-f/l-α) can be prepared from the commercially available Λ/,O-dimethylhydroxylamine hydrochloride and an activating agent (e.g., trimethylaluminum). In step 6 of Scheme 1 , the arylbenzyl group (Ar) is introduced using the desired organometallic reagent (e.g., organo lithium compound (ArLi) or organomagnesium compound (ArMgX)) in tetrahydrofuran (THF) at a temperature ranging from about -780C to about 2O0C followed by hydrolysis (upon standing in protic conditions) to the corresponding lactol (N) which may be in equilibrium with the corresponding ketone (Ni). The bridged ketal motif found in (A) and (B) can be prepared by removing the protecting groups (Pg2) using the appropriate reagents for the protecting groups employed. For example, the PMB protecting groups may be removed by treatment with trifluoroacetic acid in the presence of anisole and dichloromethane (DCM) at about O0C to about 230C (room temperature). The remaining protecting groups (Pg1) may then be removed using the appropriate chemistry for the particular protecting groups. For example, benzyl protecting groups may be removed by treating with formic acid in the presence of palladium (Pd black) in a protic solvent (e.g., ethanol/THF) at about room temperature to produce the final products (A) and (B). When R1 is CN, the use of a Lewis acid like boron trichloride at a temperature ranging from about -780C to about room temperature in a solvent like dichloromethane or 1 ,2-dichloroethane may also be used to remove benzyl protective and/or para- methoxybenzyl protective groups. When R1 is CN and R2 is (Ci-C4)alkoxy in intermdediate (l-i) or in products (A) or (B), upon treatment with a Lewis acid such as boron trichloride or boron tribomide, partial to complete de-alkylation to the corresponding phenol may occur to lead to the corresponding compound (A) or (B) where R1 is CN and R2 is OH. If this occurs, the (d- C4)alkoxy group may be re-introduced via selective alkylation using a (CrC4) alkyl iodide under mildly basic conditions, for example, potassium carbonate in acetone at a temperature ranging from about room temperature to about 56 degrees Celsius.
When R1 and/or R2 is (CrC4)alkyl-SO2- it is understood by one skilled in the art that the organometallic addition step 6 (Scheme 1 ) will be carried out on the corresponding (d- C4)alkyl-S- containing organometallic reagent. The thio-alkyl is then oxidized at a later stage to the corresponding sulfone using conventional methods known by those skilled in the art.
The compounds of the present invention may be prepared as co-crystals using any suitable method. A representative scheme for preparing such co-crystals is described in Scheme 2.

Scheme 2
In Scheme 2, wherein Me is methyl and Et is ethyl, in step 1 , 1-(5-bromo-2- chlorobenzyl)-4-ethoxybenzene is dissolved in 3:1 , toluene: tetrahydrofuran followed by cooling the resulting solution to <-70°C. To this solution is added hexyllithium while maintaining the reaction at <-65°C followed by stirring for 1 hour. (3R,4S,5R,6R)-3,4,5- ths(thmethylsilyloxy)-6-((trimethylsilyloxy)methyl)-tetrahydropyran-2-one (ll-a) is dissolved in toluene and the resulting solution is cooled to -150C. This solution is then added to the – 7O0C aryllithium solution followed by stirring for 1 hour. A solution of methanesulfonic acid in methanol is then added followed by warming to room temperature and stirring for 16 to 24 hours. The reaction is deemed complete when the α-anomer level is < 3%. The reaction is then basified by the addition of 5 M aqueous sodium hydroxide solution. The resulting salts are filtered off followed by concentration of the crude product solution. 2- methyltetrahydrofuran is added as a co-solvent and the organic phase is extracted twice with water. The organic phase is then concentrated to 4 volumes in toluene. This concentrate is then added to a 5:1 , heptane: toluene solution causing precipitate to form. The solids are collected and dried under vacuum to afford a solid.
In step 2 of Scheme 2, to (ll-b) in methylene chloride is added imidazole followed by cooling to O0C and then addition of trimethylsilylchlohde to give the persilylated product.
The reaction is warmed to room temperature and quenched by the addition of water, and the organic phase is washed with water. This crude methylene chloride solution of (ll-c) is dried over sodium sulfate and then taken on crude into the next step.
In step 3 of Scheme 2, the crude solution of (ll-c) in methylene chloride is concentrated to low volume and then the solvent is exchanged to methanol. The methanol solution of (ll-c) is cooled to O0C, then 1 mol% of potassium carbonate is added as a solution in methanol followed by stirring for 5 hours. The reaction is then quenched by addition of 1 mol% acetic acid in methanol, followed by warming to room temperature, solvent exchange to ethyl acetate, and then filtration of the minor amount of inorganic solids. The crude ethyl acetate solution of (ll-d) is taken directly into the next step.
In step 4 of Scheme 2, the crude solution of (ll-d) is concentrated to low volume, then diluted with methylene chloride and dimethylsulfoxide. Triethylamine is added followed by cooling to 1O0C and then sulfur trioxide pyridine complex is added in 3 portions as a solid at 10 minute intervals. The reaction is stirred an additional 3 hours at 1O0C before quenching with water and warming to room temperature. The phases are separated followed by washing the methylene chloride layer with aqueous ammonium chloride. The crude methylene chloride solution of (ll-e) is taken directly into the next step.
In step 5 of Scheme 2, the crude solution of (ll-e) is concentrated to low volume and then the solvent is exchanged to ethanol. Thirty equivalents of aqueous formaldehyde is added followed by warming to 550C. An aqueous solution of 2 equivalents of potassium phosphate, tribasic is added followed by stirring for 24 hours at 550C. The reaction temperature is then raised to 7O0C for an additional 12 hours. The reaction is cooled to room temperature, diluted with te/t-butyl methyl ether and brine. The phases are separated followed by solvent exchange of the organic phase to ethyl acetate. The ethyl acetate phase is washed with brine and concentrated to low volume. The crude concentrate is then purified by silica gel flash chromatography eluting with 5% methanol, 95% toluene. Product containing fractions are combined and concentrated to low volume.
Methanol is added followed by stirring until precipitation occurs. The suspension is cooled and the solids are collected and rinsed with heptane followed by drying. Product (ll-f) is isolated as a solid.
In step 6 of Scheme 2, compound (ll-f) is dissolved in 5 volumes of methylene chloride followed by the addition of 1 mol% SiliaBonc/® tosic acid and stirring for 18 hours at room temperature. The acid catalyst is filtered off and the methylene chloride solution of (ll-g) is taken directly into the next step co-crystallization procedure.
In step 7 of Scheme 2, the methylene chloride solution of (ll-g) is concentrated and then the solvent is exchanged to 2-propanol. Water is added followed by warming to 550C. An aqueous solution of L-pyroglutamic acid is added followed by cooling the resulting solution to room temperature. The solution is then seeded and granulated for 18 hours. After cooling, the solids are collected and rinsed with heptane followed by drying. Product (ll-h) is isolated as a solid.
An alternative synthesis route for compounds (A) of the present invention is depicted in Scheme 3 and described below.

Scheme 3
The synthesis of (lll-a), where R3 is an alkyl or fluoro substituted alkyl (except for the carbon adjacent to the oxygen atom) can be prepared in a similar way as described in step 1 of Scheme 2. In step 1 of Scheme 3, the primary hydroxyl group is selectively protected by an appropriate protective group. For example, a trityl group (Pg3 = Tr) can be introduced by treatment of intermediate (lll-a) with chlorotriphenylmethane in presence of a base like pyridine in a solvent like toluene, tetrahydrofuran or dichloromethane at a temperature ranging from about 0 degrees Celsius to about room temperature. Additional examples of such protective groups and experimental conditions are known by those skilled in the art and can be found in T. W. Greene, Protective Groups in Organic Synthesis. John Wiley & Sons, New York, 1991.
In step 2 of Scheme 3, the secondary hydroxyl groups can be protected by the appropriate protecting groups. For example, benzyl groups (Pg4 is Bn) can be introduced by treatment of intermediate (lll-b) with benzyl bromide or benzyl chloride in the presence of sodium hydride, potassium hydride, potassium te/t-butoxide in a solvent like tetrahydrofuran, 1 ,2-dimethoxyethane or Λ/,Λ/-dimethylformamide at a temperature ranging from about 0 degrees Celsius to about 80 degrees Celsius. Acetyl or benzoyl groups (Pg4 = Ac or Bz) may be introduced by treatment of intermediate (lll-b) with acetyl chloride, acetyl bromide or acetic anhydride or benzoyl chloride or benzoic anhydride in the presence of a base like triethylamine, Λ/,Λ/-diisopropylethylamine or 4-
(dimethylamino)pyridine in a solvent like tetrahydrofuran, 1 ,2-dimethoxyethane or dichloromethane at a temperature ranging from about 0 degrees Celsius to about 80 degrees Celsius.
In step 3 of Scheme 3, the primary hydroxyl group is deprotected to lead to intermediate (lll-d). When Pg3 is Tr, intermediate (lll-c) is treated in the presence of an acid like para-toluenesulfonic acid in a alcoholic solvent like methanol at a temperature ranging from about -20 degrees Celsius to about room temperature to provide intermediate (lll-d). Cosolvents like chloroform may be used.
In step 4 of Scheme 3, a hydroxymethylene group is introduced through a process similar to the one already described in Scheme 1 (step 1 ) and Scheme 2 (steps 4 and 5).
Other sources of formaldehyde, like paraformaldehyde in a solvent like ethanol at a temperature ranging from about room temperature to about 70 degrees Celsius in the presence of an alkali metal alkoxide can also be used in this step. When Pg4is Bn, this step provides intermediate (lll-e) and when Pg4 is Ac or Bz, this step provides intermediate (lll-f).
In step 5 of Scheme 3, intermediate (lll-e) is treated with an acid like trifluoroacetic acid or an acidic resin in a solvent like dichloromethane at a temperature ranging from about -10 degrees Celsius to about room temperature to produce intermediate (lll-g).
In step 6 of Scheme 3, the remaining protecting groups (Pg4) may then be removed using the appropriate chemistry for the particular protecting groups. For example, benzyl protecting groups may be removed by treating with formic acid in the presence of palladium (Pd black) in a protic solvent (e.g., ethanol/THF) at about room temperature to produce the final product (A).
In step 7 of Scheme 3, intermediate (lll-f) is treated with an acid like trifluoroacetic acid or an acidic resin in a solvent like dichloromethane at a temperature ranging from about -10 degrees Celsius to about room temperature to produce the final product (A). Another alternative scheme for synthesizing product (A) is depicted in Scheme 4 and described below.

Scheme 4 In step 1 of Scheme 4, intermediate (lll-a) is treated with the appropriate arylsulfonyl chloride R4SO2CI or arylsulfonic anhydride R4S(O)2OS(O)2R4 (wherein R4 is an optionally substituted aryl group, such as found in the arylsulfonyl chlorides 4-methyl-benzenesulfonyl chloride, 4-nitro-benzenesulfonyl chloride, 4-fluoro-benzenesulfonyl chloride, 2,6-dichloro- benzenesulfonyl chloride, 4-fluoro-2-methyl-benzenesulfonyl chloride, and 2,4,6-trichloro- benzenesulfonyl chloride, and in the arylsulfonic anhydride, p-toluenesulfonic anhydride) in presence of a base like pyridine, triethylamine, Λ/,Λ/-diisopropylethylamine in a solvent like tetrahydrofuran, 2-methyltetrahydrofuran at a temperature ranging from about -20 degrees Celsius to about room temperature. Some Lewis acids like zinc(ll) bromide may be used as additives. In step 2 of Scheme 4, intermediate (IV-a) is submitted to a Kornblum-type oxidation
(see, Kornblum, N., et al., Journal of The American Chemical Society, 81 , 4113 (1959)) to produce the corresponding aldehyde which may exist in equilibrium with the corresponding hydrate and/or hemiacetal form. For example intermediate (IV-a) is treated in the presence of a base like pyridine, 2,6-lutidine, 2,4,6-collidine, Λ/,Λ/-diisopropylethylamine, A- (dimethylamino)pyridine in a solvent like dimethyl sulfoxide at a temperature ranging from about room temperature to about 150 degrees Celsius. The aldehyde intermediate produced is then submitted to the aldol/Cannizzaro conditions described for step 1 (Scheme 1 ) and step 5 (Scheme 2) to produce intermediate (IV-b). In step 3 of Scheme 4, intermediate (IV-b) is treated with an acid like thfluoroacetic acid or an acidic resin in a solvent like dichloromethane at a temperature ranging from about -10 degrees Celsius to about room temperature to produce the final product (A).
When R2 is (C2-C4)alkynyl the process may be performed using Scheme 5, wherein R6 is H or (CrC2)alkyl.

Scheme 5
In step 1 of Scheme 5, which provides intermediate (V-i), the organometallic addition step is carried out in a similar way to the one described in Schemel , step 6, using the organometallic reagent derived from (V-a), where Pg5 is a suitable protective group for the hydroxyl group. For instance Pgs can be a te/t-butyldimethylsilyl group (TBS) (see
US2007/0054867 for preparation of for instance {4-[(5-bromo-2-chloro-phenyl)-methyl]- phenoxy}-te/t-butyl-dimethyl-silane).
In step 2 of Scheme 5, when Pg2 = PMB, intermediate (V-i) is treated with an acid like trifluoroacetic acid, methanesulfonic acid or an acidic resin in presence of anisole in a solvent like dichloromethane at a temperature ranging from about -10 degrees Celsius to about room temperature to produce intermediate (V-j).
In step 3 of Scheme 5, protecting groups (Pg5) and (Pg1) can be removed to provide (V-k). Typically (Pg5) is TBS and Pg1 is Bn. In this circumstance, the protecting groups are removed by sequential treatment of (V-j) with 1 ) tetrabutylammonium fluoride in a solvent like tetrahydrofuran or 2-methyltetrahydrofuran at a temperature ranging from 0 degrees
Celsius to about 40 degrees Celsius and 2) treatment with formic acid in the presence of palladium (Pd black) in a protic solvent (e.g., ethanol/THF) at about room temperature. In this sequence, the order of the 2 reactions is interchangeable.
In step 4 of Scheme 5, intermediate (V-k) is treated with N,N-bis- (trifluoromethanesulfonyl)-aniline in presence of a base like triethylamine or 4- dimethyaminopyridine in a solvent like dichloromethane or 1 ,2-dichloroethane at a temperature ranging from 0 degrees Celsius to about 40 degrees Celsius to produce intermediate (V-I).
In step 5 of Scheme 5, intermediate (V-I) is subjected to a Sonogashira-type reaction (see, Sonogashira, K. Coupling Reactions Between sp2 and sp Carbon Centers. In
Comprehensive Organic Synthesis (eds. Trost, B. M., Fleming, I.), 3, 521-549, (Pergamon, Oxford, 1991 )).

IS ERTUGLIFLOZIN
Example 4
(1 S.2S.3S.4R.5S)-5-[4-chloro-3-(4-ethoxy-benzyl)-Dhen yll- 1 -h vdroxymeth yl-6.8-dioxa- bicvclo[3.2.1loctane-2,3Λ-triol (4A) and (1S,2S,3SΛS,5S)-5-[4-chloro-3-(4-ethoxy- benzvD-phen yll- 1 -h vdroxymeth yl-6, 8-dioxa-bicvclo[3.2.1 loctane-2, 3, 4-triol (4B):

To a solution of {(2S,3S)-2,3,4-tris-benzyloxy-5-[4-chloro-3-(4-ethoxy-benzyl)-phenyl]-6,8- dioxa-bicyclo[3.2.1]oct-1-yl}-methanol (l-4k: 335 mg) in ethanol/tetrahydrofuran (10 ml_, 4/1 volume) was added successively formic acid (420 microL, 22 equivalents) and palladium black (208 mg, 4 equivalents) and the resulting mixture was stirred at room temperature. After 1 hour, additional formic acid (420 microL, 22 equivalents) and palladium black (208 mg, 4 equivalents) were added and the mixture was allowed to stir for an additional hour at room temperature. The palladium was filtered and the crude mixture obtained after evaporation of solvent was purified by HPLC preparative.
HPLC preparative: reverse phase C18 Gemini column 5 micrometer 30 x 100 mm, 40 mL/minute, gradient of acetonitrile/0.1 % formic acid : water/0.1 % formic acid; 25 to 50% of acetonitrile/0.1 % formic acid over 18 minutes; UV detection: 220 nm. The HPLC indicated a ratio of diastereomers of 1.1 :1 (4A:4B).
4A: (60 mg, 29% yield); Rt = 12.4 minutes; the fractions containing the product were concentrated under reduced pressure. The crude material was precipitated from ethyl acetate and heptane. The resulting white solid was washed with heptane 2 times and dried under reduced pressure.
MS (LCMS) 437.3 (M+H+; positive mode); 481.3 (M+HCO2 ~; negative mode). 1H NMR (400 MHz, methanol-d4) delta 7.43 (d, 1 H, J = 1.9 Hz), 7.36 (dd, 1 H, J = 8.3 and 2Hz), 7.32 (d, 1 H, J = 8.3 Hz), 7.08-7.04 (m, 2H), 6.79-6.75 (m, 2H), 4.12 (d, 1 H, J = 7.5 Hz), 4.00 (s, 2H), 3.96 (q, 2H, J = 7.0 Hz), 3.81 (d, 1 H, J = 12.5 Hz), 3.75 (dd, 1 H, J = 8.3 and 1.3 Hz), 3.65 (d, 1 H, J = 12.5 Hz), 3.63 (t, 1 H, J = 8.2 Hz), 3.57 (dd, 1 H, J = 7.5 and 1.3 Hz), 3.52 (d, 1 H, J = 8.0 Hz), 1.33 (t, 3H, J = 6.9 Hz). HRMS calculated for C22H26O7CI (M+H+) 437.1361 , found 437.1360.
4B: (30 mg, 15% yield); Rt = 13.2 minutes; the fractions containing the product were concentrated under reduced pressure. The crude material was precipitated from ethyl acetate and heptane. The resulting white solid was washed with heptane 2 times and dried under reduced pressure.
MS (LCMS) 437.3 (M+H+; positive mode) 481.3 (M+HCO2 “, negative mode). 1H NMR (400 MHz, methanol-d4) delta 7.48 (d, 1 H, J = 1.9 Hz) 7.40 (dd, 1 H, J = 8.1 and 1.9 Hz), 7.32 (d, 1 H, J = 8.3 Hz), 7.08-7.03 (m, 2H), 6.80-6.74 (m, 2H), 4.04-3.99 (m, 3H), 3.95 (q, 2H, J = 7 Hz), 3.89-3.81 (m, 4H), 3.73 (d, 1 H, J = 12.5 Hz), 3.49 (d, 1 H, J = 7.3 Hz), 1.32 (t, 3H, J = 7 Hz). HRMS calculated for C22H26O7CI (M+H+) 437.1361 , found 437.1358.
Merck & Co., Inc. and Pfizer Enter Worldwide Collaboration Agreement to Develop and Commercialize Ertugliflozin, an Investigational Medicine for Type 2 Diabetes
 ERTUGLIFLOZIN
ERTUGLIFLOZIN
Merck & Co., Inc. (NYSE: MRK), known as MSD outside the United States and Canada (“Merck”), and Pfizer Inc. (NYSE:PFE) today announced that they have entered into a worldwide (except Japan) collaboration agreement for the development and commercialization of Pfizer’s ertugliflozin (PF-04971729), an investigational oral sodium glucose cotransporter (SGLT2) inhibitor being evaluated for the treatment of type 2 diabetes. Ertugliflozin is Phase III ready, with trials expected to begin later in 2013.
“We are pleased to join forces with Merck in the battle against type 2 diabetes and the burden that it poses on global health,” said John Young, president and general manager, Pfizer Primary Care. “Through this collaboration, we believe we can build on Merck’s leadership position in diabetes care with the introduction of ertugliflozin, an innovative SGLT2 inhibitor discovered by Pfizer scientists.”
Under the terms of the agreement, Merck, through a subsidiary, and Pfizer will collaborate on the clinical development and commercialization of ertugliflozin and ertugliflozin-containing fixed-dose combinations with metformin and JANUVIA® (sitagliptin) tablets. Merck will continue to retain the rights to its existing portfolio of sitagliptin-containing products. Pfizer has received an upfront payment and milestones of $60 million and will be eligible for additional payments associated with the achievement of pre-specified future clinical, regulatory and commercial milestones. Merck and Pfizer will share potential revenues and certain costs on a 60/40 percent basis.
“Merck continues to build upon our leadership position in the oral treatment of type 2 diabetes through our own research and business development,” said Nancy Thornberry, senior vice president and Diabetes and Endocrinology franchise head, Merck Research Laboratories. “We believe ertugliflozin has the potential to complement our strong portfolio of investigational and marketed products, and we look forward to collaborating with Pfizer on its development.”
……………….
Development of an Early-Phase Bulk Enabling Route to Sodium-Dependent Glucose Cotransporter 2 Inhibitor Ertugliflozin

The development and optimization of a scalable synthesis of sodium-dependent glucose cotransporter 2 inhibitor, ertugliflozin, for the treatment of type-2 diabetes is described. Highlights of the chemistry are a concise, four-step synthesis of a structurally complex API from known intermediate 4 via persilylation–selective monodesilylation, primary alcohol oxidation, aldol-crossed-Cannizzaro reaction, and solid-phase acid-catalyzed bicyclic ketal formation. The final API was isolated as the l-pyroglutamic acid cocrystal.
1= ertugliflozin
| PF-04971729, a potent and selective inhibitor of the sodium-dependent glucose cotransporter 2, is currently in phase 2 trials for the treatment of diabetes mellitus. Inhibitory effects against the organic cation transporter 2-mediated uptake of [14C] metformin by PF- 04971729 also were very weak (IC50 900μM). The disposition of PF-04971729, an orally active selective inhibitor of the sodium-dependent glucose cotransporter 2, was studied after a single 25-mg oral dose of [14C]-PF-04971729 to healthy human subjects. The absorption of PF-04971729 in humans was rapid with a Tmax at ~ 1.0 h. Of the total radioactivity excreted in feces and urine, unchanged PF-04971729 collectively accounted for ~ 35.3% of the dose, suggestive of moderate metabolic elimination in humans. |
| References on PF-04971729: [1]. 1. Amit S. Kalgutkar, Meera Tugnait, Tong Zhu, et al.Preclinical Species and Human Disposition of PF-04971729, a Selective Inhibitor of the Sodium-Dependent Glucose cotransporter 2 and Clinical Candidate for the Treatment of Type 2 . Diabetes Mellitus Drug Metabolism and Diposition, 2011, 39 (9):. 1609-1619 Abstract (1S, 2S, 3S, 4R, 5S) -5 – [4-Chloro-3-(4-ethoxybenzyl) phenyl] -1 -hydroxymethyl-6 ,8-dioxabicyclo [3.2.1] octane-2 ,3,4-triol (PF-04971729), a potent and selective inhibitor of the sodium-dependent glucose cotransporter 2, is currently in phase 2 trials for the treatment of diabetes mellitus. This article describes the preclinical species and in vitro human disposition characteristics of PF-04971729 that were used in experiments performed to support the first-in-human study. Plasma clearance was low in rats (4.04 ml · min? 1 · kg? 1) and dogs (1.64 ml · min? 1 · kg? 1), resulting in half-lives of 4.10 and 7.63 h, respectively. Moderate to good bioavailability in rats (69%) and dogs (94%) was . observed after oral dosing The in vitro biotransformation profile of PF-04971729 in liver microsomes and cryopreserved hepatocytes from rat, dog, and human was qualitatively similar;. prominent metabolic pathways included monohydroxylation, O-deethylation, and glucuronidation No human-specific metabolites of PF-04971729 were detected in in vitro studies. Reaction phenotyping studies using recombinant enzymes indicated a role of CYP3A4/3A5, CYP2D6, and UGT1A9/2B7 in the metabolism of PF-04971729. No competitive or time-dependent inhibition of the major human cytochrome P450 enzymes was discerned with PF-04971729. Inhibitory effects against the organic cation transporter 2-mediated uptake of [14C] metformin by PF-04971729 also were very weak (IC50 =? 900 μM). Single-species allometric scaling of rat pharmacokinetics of PF-04971729 was used to predict human clearance, distribution volume, and oral bioavailability. Human pharmacokinetic predictions were consistent with the potential for a low daily dose. First-in-human studies after oral administration indicated that the human pharmacokinetics / dose predictions for PF -04971729 were in the range that is likely to yield a favorable pharmacodynamic response.. [2] … Timothy Colin Hardman, Simon William Dubrey Development and potential role of type-2 sodium-glucose transporter Inhibitors for Management of type 2 Diabetes Diabetes Ther 2011 September; 2 (3):. 133-145 Abstract There is a recognized need for new treatment options for type 2 diabetes mellitus (T2DM). Recovery of glucose from the glomerular filtrate represents an important mechanism in maintaining glucose homeostasis and represents a novel target for the management of T2DM. Recovery of glucose from the glomerular filtrate is executed principally by the type 2 sodium-glucose cotransporter (SGLT2). Inhibition of SGLT2 promotes glucose excretion and normalizes glycemia in animal models. First reports of specifically designed SGLT2 inhibitors began to appear in the second half of the 1990s. Several candidate SGLT2 inhibitors are currently under development, with four in the later stages of clinical testing. The safety profile of SGLT2 inhibitors is expected to be good, as their target is a highly specific membrane transporter expressed almost exclusively within the renal tubules. One safety concern is that of glycosuria , which could predispose patients to increased urinary tract infections. So far the reported safety profile of SGLT2 inhibitors in clinical studies appears to confirm that the class is well tolerated. Where SGLT2 inhibitors will fit in the current cascade of treatments for T2DM has yet to be established. The expected favorable safety profile and insulin-independent mechanism of action appear to support their use in combination with other antidiabetic drugs. Promotion of glucose excretion introduces the opportunity to clear calories (80-90 g [300-400 calories] of glucose per day) in patients that are generally overweight, and is expected to work synergistically with weight reduction programs. Experience will most likely lead to better understanding of which patients are likely to respond best to SGLT2 inhibitors, and under what circumstances.[3]. Zhuang Miao, Gianluca Nucci, Neeta Amin. Pharmacokinetics, Metabolism and Excretion of the Anti-Diabetic Agent Ertugliflozin (PF-04971729) in Healthy Male the Subjects. Drug Metabolism and Diposition. Abstract The Disposition of ertugliflozin (PF-04971729) , an orally active selective inhibitor of the sodium-dependent glucose cotransporter 2, was studied after a single 25-mg oral dose of [14C]-PF-04971729 to healthy human subjects. Mass balance was achieved with approximately 91% of the administered dose recovered in urine and feces. The total administered radioactivity excreted in feces and urine was 40.9% and 50.2%, respectively. The absorption of PF-04971729 in humans was rapid with a Tmax at ~ 1.0 h. Of the total radioactivity excreted in feces and urine, unchanged PF-04971729 collectively accounted for ~ 35.3% of the dose, suggestive of moderate metabolic elimination in humans. The principal biotransformation pathway involved glucuronidation of the glycoside hydroxyl groups to yield three regioisomeric metabolites M4a, M4b and M4c (~ 39.3% of the dose in urine) of which M4c was the major regioisomer (~ 31.7% of the dose). The structure of M4a and M4c were confirmed to be PF-04971729-4-O-β-and-3-O-β-glucuronide , respectively, via comparison of the HPLC retention time and mass spectra with authentic standards. A minor metabolic fate involved oxidation by cytochrome P450 to yield monohydroxylated metabolites M1 and M3 and des-ethyl PF-04971729 (M2), which accounted for ~ 5.2% of the dose in excreta. In plasma, unchanged PF-04971729 and the corresponding 4-O-β-(M4a) and 3-O-β-(M4c) glucuronides were the principal components, which accounted for 49.9, 12.2 and 24.1% of the circulating radioactivity. Overall, these data suggest that PF-04971729 is well absorbed in humans, and eliminated largely via glucuronidation.. [4] .. Tristan S. Maurer, Avijit Ghosh, Nahor Haddish-Berhane pharmacodynamic Model of Sodium-Glucose Transporter 2 (SGLT2) Inhibition: Implications for Quantitative Translational Pharmacology AAPS J. 2011; 13 (4): 576-584 Abstract Sodium-glucose co-transporter-2 (SGLT2) inhibitors are an emerging class of agents for use in the treatment of type 2 diabetes mellitus (T2DM). Inhibition of SGLT2 leads to improved glycemic control through increased urinary glucose excretion (UGE). In this study, a biologically based pharmacokinetic / pharmacodynamic (PK / PD) model of SGLT2 inhibitor-mediated UGE was developed. The derived model was used to characterize the acute PK / PD relationship of the SGLT2 inhibitor, dapagliflozin, in rats. The quantitative translational pharmacology of dapagliflozin was examined through both prospective simulation and direct modeling of mean literature data obtained for dapagliflozin in healthy subjects. Prospective simulations provided time courses of UGE that were of consistent shape to clinical observations, but were modestly biased toward under prediction. Direct modeling provided an improved characterization of the data and precise parameter estimates which were reasonably consistent with those predicted from preclinical data. Overall, these results indicate that the acute clinical pharmacology of SGLT2 inhibitors in healthy subjects can be reasonably well predicted from preclinical data through rational accounting of species differences in pharmacokinetics, physiology, and SGLT2 pharmacology. Because these data can be generated at the earliest stages of drug discovery, the proposed model is useful in the design and development of novel SGLT2 inhibitors. In addition, this model is expected to serve as a useful foundation for future efforts to understand and predict the effects of SGLT2 inhibition under chronic administration and in other patient populations.[5]. Yoojin Kim, Ambika R Babu Clinical potential of sodium-glucose cotransporter 2 Inhibitors in the Management of type 2 Diabetes Diabetes Obes Metab Syndr 2012; 5:…. 313-327 Abstract Background The Kidney plays an Important role in glucose metabolism, and has been considered a target for therapeutic intervention. The sodium-glucose cotransporter type 2 (SGLT2) mediates most of the glucose reabsorption from the proximal renal tubule. Inhibition of SGLT2 leads to glucosuria and provides a unique mechanism to lower elevated blood glucose levels in diabetes. The purpose of this review is to explore the physiology of SGLT2 and discuss several SGLT2 inhibitors which have clinical data in patients with type 2 diabetes. Methods We performed a PubMed search using the terms “SGLT2″ and “SGLT2 inhibitor” through April 10, 2012. Published articles, press releases, and abstracts presented at national and international meetings were considered. Results SGLT2 inhibitors correct a novel pathophysiological defect, have an insulin-independent action, are efficacious with glycosylated hemoglobin reduction ranging from 0.5% to 1.5%, promote weight loss, have a low incidence of hypoglycemia, complement the action of other antidiabetic agents, and can be used at any stage of diabetes. They are generally well tolerated. However, due to side effects, such as repeated urinary tract and genital infections, increased hematocrit, and decreased blood pressure, appropriate patient selection for drug initiation and close monitoring after initiation will be important. Results of ongoing clinical studies of the effect of SGLT2 inhibitors on diabetic complications and cardiovascular safety are crucial to determine the risk -benefit ratio. A recent decision by the Committee for Medicinal Products for Human Use of the European Medicines Agency has recommended approval of dapagliflozin for the treatment of type 2 diabetes as an adjunct to diet and exercise, in combination with other glucose-lowering medicinal products , including insulin, and as a monotherapy for metformin-intolerant patients. Clinical research also remains to be carried out on the long-term effects of glucosuria and other potential effects of SGLT2 inhibitors, especially in view of the observed increase in the incidence of bladder and breast cancer SGLT2 inhibitors represent a promising approach for the treatment of diabetes, and could potentially be an addition to existing therapies Keywords:.. sodium-glucose cotransporter type 2, SGLT2, inhibitors, kidney, glucosuria, oral diabetes agent, weight loss.[6]. Clinical Trials with PF-04971729 |
Example 6 Manufacturing Process for Tablets US20130137646
 |
Triphala : A Digestive Miracle


| Terminalia bellirica | |
|---|---|

|
| Terminalia chebula | |
|---|---|
_leafless_tree_at_23_Mile,_Duars,_WB_W_IMG_5905.jpg) |

Triphala (/triːˈfɑːlə/ or /triːˈfælə/; Hindi/Sanskrit: त्रिफला, triphalā [trɪˈpʰɐlaː], “three fruits”)[1] is an Ayurvedic[2] herbal rasayana formula consisting of equal parts of three myrobalans, taken without seed: Amalaki (Emblica officinalis), Bibhitaki (Terminalia bellirica), and Haritaki (Terminalia chebula).[1]
Medicinal use
In traditional Ayurvedic medicine, Triphala is used for:
- immune system stimulation[3]
- improvement of digestion[4][1]
- relief of constipation[4][1]
- gastrointestinal tract cleansing[4]
- relief of gas[1]
- treatment of diabetes[1]
- treatment of eye disease[1]
These health claims have not been yet tested in clinical trials. Even within the practice of Ayurvedic medicine, there are controversies about the composition (amlaki, haritaki and bibhitaki), preparation, and medicinal uses of Triphala.[5]
The active constituents are unknown. Triphala contains several compounds that have been proposed to be responsible for its claimed health benefits, including gallic acid, chebulagic acid, and chebulinic acid. [6][7]
Contemporary research on triphala
There is preliminary evidence that Triphala contains compounds with antioxidant properties in isolated cells and rats, however this has not yet been demonstrated in people.[6][8][9][10]
Triphala, widely used by natural Ayurvedic healers in India for thousands of years, contains 3 different fruits: Harada, Amla and Bihara. The word “Triphala”literally means “three fruits”. The combination of these three fruits cleanses the gastro-intestinal tract in a natural and gentle way. Basically our “bathroom experience” becomes a better one That is the best way I can put it!!
Why should we cleanse?
It’s always a good idea to cleanse! Get rid of toxins that build up in our bodies so that our bodies can function most efficiently and have that bright glowing skin we all crave and want! More energy and feel less bloated!
And I’m not talking about cleansing with juicing or not eating. No no, that’s a whole other conversation. I absolutely believe in still eating a healthy diet while “cleansing”/taking Triphala.
I have suggested Triphala to many clients, students and friends and all of them have seen results. You can call it a form of laxative if you’d like but this is totally safe and gentle on the body. Yes, we are all different but seems like this one might be a miracle worker and work for everyone!
Suggested use: Take one pill before bedtime. *Take on and off for a period of time OR once in a while when you feel you need it. I usually take it when I feel I need a cleanse- about one or two times a week (usually when I have consumed a bigger meal or more food than usual).
Benefits of Triphala:
- detoxify and cleanses the colon of toxins
- removes excess fats
- purifies the blood
- removes toxins from the liver
- reduces some forms of cholesterol (serum cholesterol)
- reduces high blood pressure
- high nutritional value: including high levels of vitamin C
- high in antioxidants
- strengthens hair roots and enriches hair color


The three fruits contained in Triphala are
Amalaki (Indian Gooseberry),
Haritaki (Indian Gallnut or Terminalia chebula),
and Bibhitaki (Beleric Myrobalan or Terminalia bellerica).
The prokinetic cleanser
An immensely popular Ayurvedic herbal formula,Triphala(Terminalia chebula,Terminalia bellirica and Emblica officinalis) is an effective bowel cleanser. It combines the goodness of Indian Gooseberry, Belleric Myrobalan and Chebulic Myrobalan, which work together to produce effective bowel movements.
The herbal compound provides overall support for digestion and helps ensure that the digestive tract works at optimal levels. Triphala relieves constipation and regularizes the digestive system, without disrupting the fluid-electrolyte balance in the body.
The herbs that make up Triphala are found in abundance in India.
Triphala, the well-known traditional Ayurvedic formulation, makes an excellent skin tonic. It is one of the most popular Ayurvedic medicinal herbs, prescribed by a number of Ayurvedic practitioners. Triphala literally means ‘three fruits’. The three fruits contained in Triphala are Amalaki (Indian Gooseberry), Haritaki (Indian Gallnut or Terminalia chebula), and Bibhitaki (Beleric Myrobalan or Terminalia bellerica). Since Triphala is tridoshic – equally balancing for Vata, Pitta and Kapha – it is beneficial for all skin types. Triphala nourishes the skin, both directly and indirectly. Amla (Indian gooseberry), one of the three ingredients in Triphala, is the richest known natural source of Vitamin C. Apart from the rich source of Vitamin C, Triphala also contains calcium – an important nutrient that helps enhance skin clarity and brings dull, tired skin to life.
Preparation Of Triphala Rasayana
Triphala Rasayana is usually prepared by mixing triphala with equal quantity of madhuka (mahua tree), tavakshir (East Indian arrowroot) pippali (long pepper), saindhava (long salt), and each one of the loha (iron), suvarna (gold), vacha (Acorus calamus) with either honey, ghee or sugar, in equal quantity.
Benefits Of Triphala
Triphala Rasayana is beneficial is promoting ojas, the finest product of digestion that prevents the occurrence of many diseases, creates luster and make the skin exude its natural glow and radiance.
It nourishes both the body and the mind, thereby promoting longevity of life. Therefore, Triphala Rasayana is very much beneficial for adults and children alike.
The Rasayana is especially beneficial for eyes. In case one has problems in eye sight, opting for Triphala Rasayana would be the best bet.
The Rasayana creates a balance in the cholesterol level, by removing ama from the fat tissue.
It helps in the purification of urinary tract, thereby helping the prevention of urinary tract diseases.
It also strengthens and cleanses the liver, which is one of its main functions. This ensures that the liver, one of the important parts of the body, stays healthy. It can also be said that the consumption of Rasayana prevents diseases related to the functioning of liver.
The medicine also helps the management of weight. Thus, it is beneficial for people, who want to loose weight.
It enhances the thirteen agnis (digestive fires), especially the main digestive fire in the stomach.
Triphala Rasayana is helpful in pacifying Kapha and Pitta. If taken on a regular basis, the Rasayana can be a powerful anti-aging medicine.
People suffering from skin inflammation, heat, infection, obesity will find the consumption of Triphala Rasayana as beneficial.
Diseases such as fatigue and anemia can be effectively cured by the regular consumption of Triphala Rasayana, if taken according to the prescribed doses.
- Ayurvedic pharmacopoeia committee. The Ayurvedic Formulary of India, Part I, 2nd English ed. New Delhi: Controller of Publications; 2003
- Anne McIntyre (7 September 2005). Herbal treatment of children: Western and Ayurvedic perspectives. Elsevier Health Sciences. pp. 278–. ISBN 9780750651745. Retrieved 24 July 2010.
- Juss SS. Triphala – the wonder drug. Indian Med Gaz 1997;131:94-6.
- Nadkarni AK. Indian Materia Medica. 3rd ed. Mumbai: Popular Press; 1976. p. 1308-15.
- Harbans Singh Puri (2003). Rasayana: ayurvedic herbs for longevity and rejuvenation. CRC Press. pp. 30–. ISBN 9780415284899. Retrieved 24 July 2010.
- Reddy TC, Aparoy P, Babu NK, Kalangi SK, Reddanna P (May 2010). “Kinetics and Docking Studies of a COX-2 Inhibitor Isolated from Terminalia bellerica Fruits”. Protein Pept Lett. PMID 20441561.
- Pawar V, Lahorkar P, Anantha Narayana DB. Development of a RP-HPLC method for analysis of Triphala curna and its applicability to test variations in Triphala curna preparations. Indian J Pharm Sci [serial online] 2009 [cited 2010 Aug 1];71:382-6. Available from:http://www.ijpsonline.com/text.asp?2009/71/4/382/57286
- Mahesh R, Bhuvana S, Begum VM (August 2009). “Effect of Terminalia chebula aqueous extract on oxidative stress and antioxidant status in the liver and kidney of young and aged rats”. Cell Biochem. Funct. 27 (6): 358–63. doi:10.1002/cbf.1581. PMID 19548245.
- Sandhya T, Lathika KM, Pandey BN, et al. (October 2006). “Protection against radiation oxidative damage in mice by Triphala”. Mutat. Res. 609 (1): 17–25.doi:10.1016/j.mrgentox.2006.05.006. PMID 16860592.
- Srikumar R, Parthasarathy NJ, Manikandan S, Narayanan GS, Sheeladevi R (February 2006). “Effect of Triphala on oxidative stress and on cell-mediated immune response against noise stress in rats”. Mol. Cell. Biochem. 283 (1-2): 67–74. doi:10.1007/s11010-006-2271-0.PMID 16444587.

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
web link




