
Home » 2013 (Page 60)
Yearly Archives: 2013
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Pfizer’s Xeljanz approved in Switzerland, first European country to approve the drug
July 15 2013 | By Márcio Barra

Pfizer just issued a press release where it reports that Switzerland’s SwissMedic has approved its rheumatoid arthritis treatment Xeljanz (tofacitinib citrate) for sale. This makes Switzerland the first European country to approve Xeljanz for adult patients with moderate-to-severe active RA who had an inadequate response or intolerance to the antirheumatic agent methotrexate.
View original post 273 more words
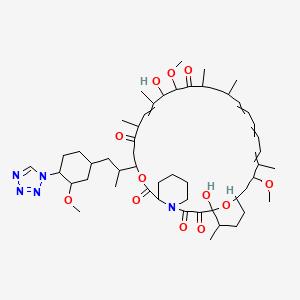
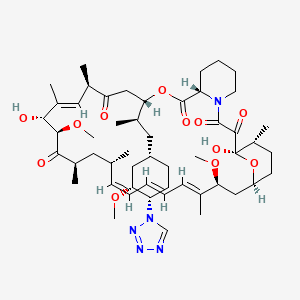
Zotarolimus, ABT 578

Zotarolimus
221877-54-9 CAS
A 179578; ABT 578; Resolute; 42-(1-Tetrazolyl)rapamycin; (42S)-42-Deoxy-42-(1H-tetrazol-1-yl)rapamycin
| Molecular Formula: C52H79N5O12 |
| Molecular Weight: 966.21 |
A tetrazole-containing Rapamycin analog as immunomodulator and useful in the treatment of restenosis and immune and autoimmune diseases.
(3S,6R,7E,9R,10R,12R,14S,15E,17E,19E,21S,23S,26R,27R,34aS)-9,27-dihydroxy-10,21-dimethoxy-3-{(1R)-2-[(1S,3R,4S)-3-methoxy-4-(1H-tetrazol-1-yl)cyclohexyl]-1-methylethyl)-6,8,12,14,20,26-hexamethyl-4,9,10,12,13,14,21,22,23,24,25,26,27,32,33,34,34a-heptadecahydro-3H-23,27-epoxypyrido[2,1-c][1,4]oxazacyclohentriacontine-1,5,11,28,29(6H,31H)-pentone, cas no 221877-54-9
zotarolimus in U.S. Patent Nos. 6,015,815 and 6,329,386 , and PCT Application No. WO 1999/015530
Zotarolimus (INN, codenamed ABT-578) is an immunosuppressant. It is a semi-synthetic derivative of rapamycin. It was designed for use in stents with phosphorylcholine as a carrier. Coronary stents reduce early complications and improve late clinical outcomes in patients needing interventional cardiology.[1] The first human coronary stent implantation was first performed in 1986 by Puel et al.[1][2] However, there are complications associated with stent use, development of thrombosis which impedes the efficiency of coronary stents, haemorrhagic and restenosis complications are problems associated with stents.[1]
These complications have prompted the development of drug-eluting stents. Stents are bound by a membrane consisting of polymers which not only slowly release zotarolimus and its derivatives into the surrounding tissues but also do not invoke an inflammatory response by the body.

Medtronic are using zotarolimus as the anti-proliferative agent in the polymer coating of their Endeavor and Resolute products.[3]
The inherent growth inhibitory properties of many anti-cancer agents make these drugs ideal candidates for the prevention of restenosis. However, these same properties are often associated with cytotoxicity at doses which block cell proliferation. Therefore, the unique cytostatic nature of the immunosuppressant rapamycin was the basis for the development of zotarolimus by Johnson and Johnson. Rapamycin was originally approved for the prevention of renal transplant rejection in 1999. More recently, Abbott Laboratories developed a compound from the same class, zotarolimus (formerly ABT-578), as the first cytostatic agent to be used solely for delivery from drug-eluting stents to prevent restenosis.[4]
Drug-eluting stents
Drug-eluting stents have revolutionized the field of interventional cardiology and have provided a significant innovation for preventing coronary artery restenosis. Polymer coatings that deliver anti-proliferative drugs to the vessel wall are key components of these revolutionary medical devices. The development of stents which elute the potent anti-proliferative agent, zotarolimus, from a synthetic phosphorylcholine-based polymer known for its biocompatible profile. Zotarolimus is the first drug developed specifically for local delivery from stents for the prevention of restenosis and has been tested extensively to support this indication. Clinical experience with the PC polymer is also extensive, since more than 120,000 patients have been implanted to date with stents containing this non-thrombogenic coating.[4]
Structure and properties

Zotarolimus is a analog made by substituting a tetrazole ring in place of the native hydroxyl group at position 42 in rapamycin that is isolated and purified as a natural product from fermentation. This site of modification was found to be the most tolerant position to introduce novel structural changes without impairing biologic activity. The compound is extremely lipophilic, with a very high octanol:water partition coefficient, and therefore has limited water solubility. These properties are highly advantageous for designing a drug-loaded stent containing zotarolimus in order to obtain a slow sustained release of drug from the stent directly into the wall of coronary vessels. The poor water solubility prevents rapid release into the circulation, since elution of drug from the stent will be partly dissolution rate-limited. The slow rate of release and subsequent diffusion of the molecule facilitates the maintenance of therapeutic drug levels eluting from the stent. In addition, its lipophilic character favors crossing cell membranes to inhibit neointimal proliferation of target tissue. The octanol:water partition coefficients of a number of compounds, recently obtained in a comparative study, indicate that zotarolimus is the most lipophilic of all DES drugs [4]
Stents are used to treat serious decreases in vessel or duct diameter due to a variety of diseases and conditions, especially atherosclerotic diseases, and are often used after angioplasty. While frequently used in arteries, stents are also used in other structures, including veins, bile ducts, esophagus, trachea, large bronchi, ureters, and urethras. Stents are the innovation of the English dentist Charles Stent (1845-1901).
While effective in treating deleterious lumen narrowing, vascular stents in an instance of medical irony, also risk re-creating the condition that they were used to treat. Stents can incur the development of thick endothelial tissue inside the lumen—the neointima. While the degree of development varies, the neointima can grow to occlude the vessel lumen, a type of restenosis.



Previous Syntheses of Zotarolimus
Mollison presented several methods to generate zotarolimus from sirolimus (Mollison, 2000). For example, C-40 hydroxyl of sirolimus is activated with the formation of triflate, and the triflate is then purified by column chromatography. During triflate purification, some of the activated intermediate reverts to sirolimus and its epimer, epi-sirolimus, due to presence of the water during chromatography. The purified triflate is then reacted in a second step with tetrazole to produce the 40-epi-tetrazole derivative of sirolimus, that is, zotarolimus. The crude product is then purified by column chromatography. However, even with this purification, the end product could contain sirolimus and epi-sirolimus impurities.
ISOMERS
ABT-578 [40-epi-(1-tetrazolyl)-rapamycin], known better today as zotarolimus, is a semi-synthetic macrolide triene antibiotic derived from rapamycin. Zotarolimus’ structure is shown in Formula D.

………………………

A representative procedure is shown in Scheme 1.

As shown in Scheme 1, conversion of the C-42 hydroxyl of rapamycin to a trifluoromethanesulfonate or fluorosulfonate leaving group provided A. Displacement of the leaving group with tetrazole in the presence of a hindered, non-nucleophilic base, such as 2,6-lutidine, or, preferably, diisopropylethyl amine provided epimers B and C, which were separated and purified by flash column chromatography.
Synthetic Methods
The foregoing may be better understood by reference to the following examples which illustrate the methods by which the compounds of the invention may be prepared and are not intended to limit the scope of the invention as defined in the appended claims.
Example 1 42-Epi-(tetrazolyl)-rapamycin (less polar isomer) Example 1AA solution of rapamycin (100 mg, 0.11 mmol) in dichloromethane (0.6 mL) at −78° C. under a nitrogen atmosphere was treated sequentially with 2,6-lutidine (53 uL, 0.46 mmol, 4.3 eq.) and trifluoromethanesulfonic anhydride (37 uL, 0.22 mmol), and stirred thereafter for 15 minutes, warmed to room temperature and eluted through a pad of silica gel (6 mL) with diethyl ether. Fractions containing the triflate were pooled and concentrated to provide the designated compound as an amber foam.
Example 1B 42-Epi-(tetrazolyl)-rapamycin (less polar isomer)A solution of Example 1A in isopropyl acetate (0.3 mL) was treated sequentially with diisopropylethylamine (87 L, 0.5 mmol) and 1H-tetrazole (35 mg, 0.5 mmol), and thereafter stirred for 18 hours. This mixture was partitioned between water (10 mL) and ether (10 mL). The organics were washed with brine (10 mL) and dried (Na2SO4). Concentration of the organics provided a sticky yellow solid which was purified by chromatography on silica gel (3.5 g, 70-230 mesh) eluting with hexane (10 mL), hexane:ether (4:1(10 mL), 3:1(10 mL), 2:1(10 mL), 1:1(10 mL)), ether (30 mL), hexane:acetone (1:1(30 mL)). One of the isomers was collected in the ether fractions.
MS (ESI) m/e 966 (M)−;
Example 2 42-Epi-(tetrazolyl)-rapamycin (more polar isomer) Example 2A 42-Epi-(tetrazolyl)-rapamycin (more polar isomer)Collection of the slower moving band from the chromatography column using the hexane:acetone (1:1) mobile phase in Example 1B provided the designated compound.
MS (ESI) m/e 966 (M)−.
………………………………………………….

sirolimus (commercially available or produced as described ((Paiva et al., 1991; Sehgal et al., 1975; Vezina et al., 1975) is dissolved in DCM:toluene (such as 1:2) 100. The reaction mixture is concentrated to dryness 105, and the azeo-drying process 105 is repeated 1-5 times more, more preferably 2-4 times, most preferably twice, preferably with DCM:toluene. The resulting foamy solid is dissolved in IPAc 110, and then 2,6-Lutidine is added 115. The solution is cooled to −30° C. 115. Triflic anhydride is then slowly added to the solution 115. After stirring the reaction mixture, the solution is filtered under nitrogen. The recovered salts 120 are washed with IPAc 125.

To the salts is added 1-H-tetrazole and DIEA 130. The reaction mixture is stirred at room temperature (e.g., 22-25° C.) 135and then concentrated. The crude reaction mixture is purified, using for example, a silica gel column and using, e.g., 1:1 THF:heptane to elute 140. The fractions are monitored for the N-1 isomer (which elutes more slowly than the N-2 isomer), pooled and concentrated, forming an oil. The oil is dissolved in minimum DCM and the solution loaded on a silica gel column packed in, for example, 65:35 heptane:acetone 145. The column is eluted with, for example, 65:35 heptane:acetone, the fractions monitored for the pure product, pooled and concentrated 150.


The purified product is then dissolved in t-BME, and then n-heptane is slowly added to form a precipitate while vigorously stirring the solution 150. The precipitated solids are stirred at 5-10° C., filtered, washed again with heptane, and dried on the funnel with nitrogen. The product is dissolved in acetone and treated with BHT 155. The solution is concentrated, dissolved in acetone, and then concentrated to dryness. The product is then dried under vacuum at 47° C. 160.
EXAMPLES
Example 1 Dichloromethane-Toluene Isopropylacetate One-Pot Process with Filtration (1)
In this example, zotarolimus was prepared from rapamycin in a one-pot process using dichloromethane, toluene and isopropylacetate; the preparation was then purified, concentrated, and dried. The purified product was then characterized by its 1H, 13C NMR resonances from COSY, ROESY, TOCSY, HSQC, and HMBC spectra.
Rapamycin (10 g) was dissolved in dichloromethane (DCM, 25 ml) and toluene (50 ml). The reaction mixture was concentrated to dryness. This azeo-drying process was repeated twice with DCM/toluene. The foamy solid was dissolved in isopropylacetate (IPAc, 65 ml), and 2,6-Lutidine (3.2 ml) was added. The solution was cooled to −30° C. acetonitrile-dry ice bath, and triflic anhydride (2.8 ml) was added slowly in 10 minutes. The reaction mixture was stirred for 30 minutes, and then filtered under nitrogen atmosphere. The salts were washed with IPAc (10 ml). 1-H-tetrazole (2.3 g), followed by diisopropylethylamine (DIEA, 7.4 ml) were added. The reaction mixture was stirred for 6 hours at room temperature, and then concentrated. The crude reaction mixture was purified on a silica gel column (350 g) eluting with 1:1 THF/heptane. The fractions containing product that eluted later (predominantly N-1 isomer) were collected and concentrated. The concentrated oil was dissolved in minimum DCM and loaded on a silica gel column packed in 65:35 heptane:acetone. The column was eluted with 65:35 heptane:acetone, and fractions containing pure product were concentrated.
The purified product was then dissolved in t-butylmethyl ether (t-BME, 13.5 g), and n-heptane (53 g) was added slowly with vigorous stirring. The precipitated solids were stirred at 5-10° C. for 2 hours, filtered, washed with heptane and dried on the funnel with nitrogen to give 3.2 g wet product. The solids (1.0 g) were dissolved in acetone (10 ml) and treated with 2,6-di-tert-butyl-4-ethylphenol (DEP, 0.2%). The solution was concentrated, dissolved in acetone (10 ml) and concentrated to dryness. The product was dried under vacuum for 18 hours at 47° C., yielding 0.83 g of zotarolimus. The product was characterized by its 1H, 13C NMR resonances from its COSY, ROESY, TOCSY, HSQC, and HMBC spectra.
1H-NMR (DMSO-d6, position in bracket): ppm 0.73 (Me, 43); 0.81 (Me, 49); 0.84 (Me, 46); 0.89 (Me, 48); 0.98 (Me, 45); 1.41, 1.05 (CH2, 24); 1.18, 1.10 (CH2, 36); 1.52 (CH, 37); 1.53 (CH2, 12 & 42); 1.59, 1.30 (CH2, 5); 1.41, 1.67 (CH2, 4); 1.11, 1.73 (CH2, 38); 1.21, 1.83 (CH2, 15); 1.21, 1.83 (CH2, 13); 1.62 (Me, 44); 1.73 (Me, 47); 1.76 (CH, 35); 1.60, 2.09 (CH2, 3); 1.93, 2.21 (CH2, 41); 2.05 (CH, 11); 2.22 (CH, 23); 2.47 (CH, 25); 2.40, 2.77 (CH2, 33); 3.06 (OCH3, 50); 3.16 (OCH3, 51); 3.22, 3.44 (CH2, 6); 3.29 (OCH2, 52); 3.29 (CH, 31); 3.60 (CH, 39), 3.62 (CH, 16); 3.89 (CH, 27); 4.01 (CH, 14); 4.02 (CH, 28); 4.95 (CH, 2); 5.02 (CH, 34); 5.10 (═CH, 30); 5.17 (CH, 40); 5.24 (OH, 28); 5.46 (═CH, 22); 6.09 (═CH, 18); 6.15 (═CH, 21); 6.21 (═CH, 20); 6.42 (═CH, 19); 6.42 (OH, 10), 9.30 (CH, 53).
13C NMR (DMSO-d6, position in bracket): ppm 10.4 (Me, 44); 13.1 (Me, 47); 13.6 (Me, 46); 14.5 (Me, 49); 15.5 (Me, 43 & 48); 20.3 (CH2, 4); 21.6 (Me, 45); 24.4 (CH2, 4); 26.2 (CH2, 12); 26.4 (CH2, 3); 26.8 (CH2, 41); 27.2 (CH2, 42); 29.6 (CH2, 13); 31.6 (CH2, 38), 31.7 (CH, 37); 32.9 (CH, 35); 34.8 (CH, 11); 35.2 (CH, 23); 38.2 (CH2, 36); 39.1 (CH, 25); 39.4 (CH2, 33); 39.6 (CH2, 24), 40.0 (CH2, 15); 43.4 (CH2, 6); 45.2 (CH, 31); 50.6 (CH, 2); 55.4 (OCH3, 50); 55.8 (OCH3, 52); 57.0 (OCH3, 52); 55.9 (CH, 40); 66.2 (CH, 14); 73.4 (CH, 34); 75.6 (CH, 28); 77.4 (CH, 39); 82.3 (CH, 16); 85.7 (CH, 27); 99.0 (CH, 10); 125.3 (═CH, 30); 127.0 (═CH, 18 & 19); 130.4 (═CH, 21); 132.2 (═CH, 20); 137.2 (═CMe, 29); 137.7 (═CMe, 17); 139.2 (═CH, 22); 144.6 (CH, 53); 167.0 (C═O, 8); 169.1 (C═O, 1); 199.0 (C═O, 9); 207.5 (C═O, 32); 210.7 (C═O, 26).
Example 2 Dichloromethane-Isopropylacetate One-Pot Process (2)
In this example, zotarolimus was prepared from rapamycin in a one-pot process using dichloromethane and isopropylacetate. The compound was then purified, concentrated, and dried.
Rapamycin (10 g) was dissolved in dichloromethane (DCM, 100 g). 2,6-Lutidine (2.92 g) was added. The solution was cooled to −30° C. in acetonitrile-dry ice bath, and triflic anhydride (4.62 g) was added slowly in 10 minutes. The reaction mixture was stirred for 20 minutes, and then warmed to 10° C. within 15 minutes. The reaction solution was then concentrated. The residue was dissolved in IPAc (55 g). 1-H-tetrazole (2.68 g), followed by diisopropylethylamine (DIEA, 7.08 g) were then added. The reaction mixture was stirred for 6 hours at room temperature and then concentrated. The crude reaction mixture was purified on a silica gel column (360 g), eluting with 1:1 THF:heptane. The fractions containing product that eluted later (principally N-1) were collected and concentrated. The concentrated oil was dissolved in minimum DCM and loaded on a silica gel column (180 g) that was packed in 65:35 heptane:acetone. The column was then eluted with 65:35 heptane:acetone, and fractions containing pure product were concentrated.
The purified product was dissolved in t-butylmethyl ether (t-BME, 23 g) and added slowly to n-heptane (80 g) with vigorous stirring. The precipitated solids were stirred at 5-10° C. for not longer than 1 hour, filtered, washed with heptane and dried on the funnel with nitrogen. BHT (0.015 g) was added to the solids. The solids were dissolved in acetone (20 g), passed through a filter, and concentrated. The residue was treated with acetone two times (20 g), and concentrated each time to dryness. The product was then dried under vacuum for 18 h at not more than 50° C. to give 2.9 g of zotarolimus.
Example 3 Dichloromethane One Pot Process (3)
In this example, zotarolimus was prepared from rapamycin in a one-pot process using dichloromethane. The compound was then purified, concentrated, and dried as described in Example 2.
Rapamycin (7.5 g) was dissolved in DCM (30 g). 2,6-Lutidine (1.76 g) was added. The solution was cooled to −30° C. in acetonitrile-dry ice bath, and triflic anhydride (2.89 g) was added slowly in 10 minutes. The reaction mixture was stirred for 20 minutes, and then assayed for the presence of rapamycin to determine consumption in the reaction. 1-H-tetrazole (1.44 g), followed by DIEA (5.29 g) was added. The reaction mixture was stirred for 6 hours at room temperature, and then directly loaded on a silica gel (270 g) column prepared in 1:1 THF:n-heptane (v/v). The crude reaction mixture was purified with 1:1 THF:n-heptane. The fractions containing product that elute later were collected and concentrated. The concentrated solids were dissolved in minimum DCM and loaded on a silica gel column (135 g) packed in 70:30 n-heptane:acetone. The column was eluted with 70:30 n-heptane:acetone, and fractions containing pure product, as identified by thin-layer chromatography (TLC), were concentrated.
The purified product was dissolved in t-BME (9 g), and added slowly to n-heptane (36 g) with vigorous stirring at 10±10° C. The precipitated solids were stirred at 5-10° C. for not longer than 1 hour, filtered, washed with n-heptane and dried on the funnel with nitrogen. BHT (0.006 g) was added to the solids. The solids were dissolved in acetone (20 g), passed through a filter, and concentrated. The residue was treated with acetone twice (20 g each) and concentrated each time to dryness. The product was dried under vacuum for not longer than 18 hours at not more than 50° C. to give 2.5 g of zotarolimus.
The above process, when carried out with rapamycin presence of 2,6-di-tert-butylpyridine or 2,4,6-collidine (2,3,5-trimethylpyridine) as a non-nucleophilic in step 1a gave zotarolimus of acceptable purity, but a lower yield.
Example 4 High-Pressure Liquid Chromatography HPLC Purification of Zotarolimus Prepared by the One-Pot Synthesis Method
In this example, zotarolimus was made from rapamycin using a one-pot synthesis method of the invention (using DCM), and then subjected to an additional round of purification using HPLC.
Rapamycin (3.75 g) was dissolved in dichloromethane (DCM, 15 g). 2,6-Lutidine (0.88 g) was then added. The solution was cooled to −30° C. in acetonitrile-dry ice bath, and triflic anhydride (1.45 g) was added slowly in 10 minutes. The reaction mixture was stirred for 20 minutes, and then 1-H-tetrazole (0.72 g), followed by DIEA (2.65 g) was added. The reaction mixture was stirred for 6 hours at 25° C., and then directly loaded on a silica gel (115 g) column prepared in 70:30 n-heptane:acetone. The crude reaction mixture was purified with 70:30 n-heptane:acetone. The fractions containing product were collected, and concentrated.
The concentrated solids were dissolved in acetonitrile-water and loaded on a C-18 TechniKrom column (5 cm×25 cm), and eluted with 64:36 acetonitrile-water containing 0.1% BHT. Fractions were analyzed by reverse phase (RP)—HPLC, and product fractions pooled and concentrated to remove acetonitrile. The product was extracted with ethyl acetate or isopropyl acetate, dried (sodium sulfate) and concentrated.
The purified product was dissolved in t-BME (4.5 g), and added slowly to n-heptane (18 g) with vigorous stirring at −10° C. The precipitated solids were stirred at 5-10° C. for not longer than 1 hour, filtered, washed with n-heptane and dried on the funnel with nitrogen. BHT (0.005 g) was added to the solids. The solids were dissolved in acetone (20 g), passed through a filter, and concentrated. The residue was treated with acetone twice (20 g), and concentrated each time to dryness. The product was dried under vacuum for not longer than 18 hours at not more than 50° C. to give 1.2 g of high quality zotarolimus.

- Braunwald E, Zipes D, Libby P, ed. (2001). Heart diseases: a textbook of cardiovascular disease (6th ed.). Philadelphia: Saunders Elsevier.
- Sigwart, U; Puel, J; Mirkovitch, V; Joffre, F; Kappenberger, L (1987). “Intravascular stents to prevent occlusion and restenosis after transluminal angioplasty”. The New England journal of medicine 316 (12): 701–6. doi:10.1056/NEJM198703193161201. PMID 2950322.
- “Medtronic Receives FDA Approval for Endeavor Zotarolimus-Eluting Coronary Stent System”.
- Burke, Sandra E.; Kuntz, Richard E.; Schwartz, Lewis B. (2006). “Zotarolimus (ABT-578) eluting stents”. Advanced Drug Delivery Reviews 58 (3): 437–46.doi:10.1016/j.addr.2006.01.021. PMID 16581153.
- Heitman, J; Movva, NR; Hall, MN (1991). “Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast”. Science 253 (5022): 905–9. PMID 1715094.

The FDA has approved the zotarolimus-eluting stent (Medtronic).
|
3-7-2012
|
ASSAY FOR IMMUNOSUPPRESSANT DRUGS
|
|
|
3-7-2012
|
ONE POT SYNTHESIS OF TETRAZOLE DERIVATIVES OF RAPAMYCIN
|
|
|
7-15-2011
|
NON-DENATURING LYSIS REAGENT
|
|
|
4-22-2011
|
IMMUNOSUPPRESSANT DRUG EXTRACTION REAGENT FOR IMMUNOASSAYS
|
|
|
3-30-2011
|
NON-DENATURING LYSIS REAGENT
|
|
|
10-27-2010
|
METHODS OF MANUFACTURING CRYSTALLINE FORMS OF RAPAMYCIN ANALOGS
|
|
|
10-13-2010
|
CRYSTALLINE FORMS OF RAPAMYCIN ANALOGS
|
|
|
4-21-2010
|
One pot synthesis of tetrazole derivatives of rapamycin
|
|
|
10-16-2009
|
Heparin Prodrugs and Drug Delivery Stents Formed Therefrom
|
|
|
2-20-2009
|
Medical Devices Containing Rapamycin Analogs
|
|
2-20-2009
|
Medical Devices Containing Rapamycin Analogs
|
|
|
11-26-2008
|
Medical devices containing rapamycin analogs
|
|
|
11-21-2008
|
CASCADE SYSTEM
|
|
|
9-5-2008
|
Method Of Treating Disorders Using Compositions Comprising Zotarolimus And Paclitaxel
|
|
|
7-25-2008
|
Medical Devices Containing Rapamycin Analogs
|
|
|
7-16-2008
|
Methods of administering tetrazole-containing rapamycin analogs with other therapeutic substances using medical devices
|
|
|
6-27-2008
|
Medical Devices Containing Rapamycin Analogs
|
|
|
9-8-2006
|
COMPOSITIONS AND METHODS OF ADMINISTERING RAPAMYCIN ANALOGS USING MEDICAL DEVICES FOR LONG-TERM EFFICACY
|
| WO2001087372A1 * | Apr 25, 2001 | Nov 22, 2001 | Cordis Corp | Drug combinations useful for prevention of restenosis |
| EP1826211A1 * | Feb 20, 2007 | Aug 29, 2007 | Cordis Corporation | Isomers and 42-Epimers of rapamycin alkyl ether analogs, methods of making and using the same |
| US5151413 * | Nov 6, 1991 | Sep 29, 1992 | American Home Products Corporation | Rapamycin acetals as immunosuppressant and antifungal agents |
| US5362718 * | Apr 18, 1994 | Nov 8, 1994 | American Home Products Corporation | Rapamycin hydroxyesters |
| US7193078 | Mar 1, 2005 | Mar 20, 2007 | Terumo Kabushiki Kaisha | Process for production of O-alkylated rapamycin derivatives |
| US7220755 | Nov 12, 2003 | May 22, 2007 | Biosensors International Group, Ltd. | 42-O-alkoxyalkyl rapamycin derivatives and compositions comprising same |
| US7279571 | Dec 1, 2005 | Oct 9, 2007 | Teva Gyógyszergyár Zártkörüen Müködö Részvénytársaság | Methods of preparing pimecrolimus |
| US7812155 | Nov 26, 2007 | Oct 12, 2010 | Terumo Kabushiki Kaisha | Process for preparing an o-alkylated rapamycin derivative and o-alkylated rapamycin derivative |
| US7872122 | May 8, 2009 | Jan 18, 2011 | Chunghwa Chemical Synthesis & Biotech Co., Ltd. | Process for making Biolimus A9 |
| US20050101624 * | Nov 12, 2003 | May 12, 2005 | Betts Ronald E. | 42-O-alkoxyalkyl rapamycin derivatives and compositions comprising same |
| US20090209572 | Nov 19, 2008 | Aug 20, 2009 | Biotica Technology Limited | 36-Des(3-Methoxy-4-Hydroxycyclohexyl) 36-(3-Hydroxycycloheptyl) Derivatives of Rapamycin for the Treatment of Cancer and Other Disorders |
| US20100204466 | Feb 23, 2010 | Aug 12, 2010 | Abbott Laboratories | One pot synthesis of tetrazole derivatives of rapamycin |
| US20100249415 | Mar 29, 2010 | Sep 30, 2010 | Kwang-Chung Lee | Process for preparation of temsirolimus |
READ
ANONYMOUS: “Randomised comparison of zotarolimus eluting and sirolimus-eluting stents in patients with coronary artery disease (ENDEAVOUR III)” JOURNAL OF AMERICAN COLLEGE OF CARDIOLOGY, vol. 46, no. 11, 6 December 2005 (2005-12-06), pages CS5-CS6, XP009089338
Cancer is just as deadly as it was 50 years ago. Here’s why that’s about to change.

Why haven’t we cured cancer yet? It seems like almost every day, we hear about another miraculous advance in cancer treatment. Drugs that cause tumors to shrink, gene therapies, and even a possible vaccine. And yet, our loved ones keep dying of cancer.
We spoke to cancer experts to find out why the death rate from cancer hasn’t changed in the past 50 years — and we learned how genetic therapies could transform cancer treatments tomorrow.
Top image: Juan Gaertner/Shutterstock.com
Belinostat (PXD101)

Belinostat (PXD101)
SPECTRUM
Tiny Biotech With Three Cancer Drugs Is More Alluring Takeover Bet Now
Forbes
The drug is one of Spectrum’s two drugs undergoing phase 3 clinical trials. Allergan paid Spectrum $41.5 million and will make additional payments of up to $304 million based on achieving certain milestones. So far, Raj Shrotriya, Spectrum’s chairman, …
Belinostat (PXD101) is experimental drug candidate under development byTopoTarget for the treatment of hematological malignancies and solid tumors. It is a histone deacetylase inhibitor.[1]
In 2007 preliminary results were released from the Phase II clinical trial of intravenous belinostat in combination with carboplatin and paclitaxel for relapsedovarian cancer.[2] Final results in late 2009 of a phase II trial for T cell lymphomawere encouraging.[3] Belinostat has been granted orphan drug and fast trackdesignation by the FDA.[4]

- Plumb, Jane A.; Finn, Paul W.; Williams, Robert J.; Bandara, Morwenna J.; Romero, M. Rosario; Watkins, Claire J.; La Thangue, Nicholas B.; Brown, Robert (2003). “Pharmacodynamic Response and Inhibition of Growth of Human Tumor Xenografts by the Novel Histone Deacetylase Inhibitor PXD101”. Molecular Cancer Therapeutics 2 (8): 721–728. PMID 12939461.
- “CuraGen Corporation (CRGN) and TopoTarget A/S Announce Presentation of Belinostat Clinical Trial Results at AACR-NCI-EORTC International Conference”. October 2007.
- Final Results of a Phase II Trial of Belinostat (PXD101) in Patients with Recurrent or Refractory Peripheral or Cutaneous T-Cell Lymphoma, December 2009
- “Spectrum adds to cancer pipeline with $350M deal.”. February 2010.
SEE COMPILATION ON SIMILAR COMPOUNDS AT …………..http://drugsynthesisint.blogspot.in/p/nostat-series.html
APAZIQUONE

APAZIQUONE
Apaziquone (EOquin[1]) is an indolequinone that is a bioreductive prodrug and a chemical analog of the older chemotherapeutic agent mitomycin C. In hypoxic cells, such as those on the inner surface of the urinary bladder, apaziquone is converted to active metabolites by intracellular reductases. The active metabolites alkylate DNA and lead to apoptotic cell death.[2] This activity is preferentially expressed in neoplastic cells.

Cystoscopic appearance of tumors in the bladder.
After administration of apaziquone directly into the urinary bladder, the drug and its active metabolite were not detected in plasma, and there were no systemic side effects[3][4]

Bladder Cancer
Apaziquone has been applied in clinical studies sponsored by Spectrum Pharmaceuticals and Allergan, Inc. for the treatment of superficial (non-muscle invasive) bladder cancer.[3] Approximately 70% of all newly diagnosed patients with bladder cancer have non-muscle invasive bladder cancer and over one million patients in the United States and Europe are affected by the disease. The US Food and Drug Administration (FDA) has granted Fast Track review status to apaziquone for this indication.[5]
-
“UvA researcher develops new bladder cancer medication”. University of Amsterdam. 25 Jul 2007.
- NCI. “apaziquone”. Archived from the original on 9 May 2009. Retrieved 2009-06-07.
- Puri R, Palit V, Loadman PM, et al. (October 2006). “Phase I/II pilot study of intravesical apaziquone (EO9) for superficial bladder cancer”. J. Urol. 176 (4 Pt 1): 1344–8. doi:10.1016/j.juro.2006.06.047. PMID 16952628.
- Hendricksen K, Gleason D, Young JM, et al. (July 2008). “Safety and side effects of immediate instillation of apaziquone following transurethral resection in patients with nonmuscle invasive bladder cancer”. J. Urol. 180 (1): 116–20. doi:10.1016/j.juro.2008.03.031. PMID 18485407.
- “FDA Designates Fast Track Status For Apaziquone (EOquin) For Bladder Cancer”. Medical News Today. 22 Jul 2009.
Spectrum Pharmaceuticals CLICK HERE

Cannabis-Linked Cell Receptor Might Help Prevent Colon Cancer

The study was published in the Aug. 1 issue of the journal Cancer Research.
A cannabinoid receptor lying on the surface of cells may help suppress colorectal cancer, say U.S. researchers. When the receptor is turned off, tumor growth is switched on. Cannabinoids are compounds related to the tetrahydrocannabinol (THC) found in the cannabis plant.

It’s already known that the receptor, CB1, plays a role in relieving pain and nausea, elevating mood and stimulating appetite by serving as a docking station for the cannabinoid group of signaling molecules. This study suggests that CB1 may offer a new path for cancer prevention or treatment.
In the study of human colorectal tumor specimens, the researchers also found that the drug decitabine can restore CB1 expression.In addition, mice those are prone to developing intestinal tumors and also have functioning CB1 receptors developed fewer and smaller tumors when treated with a drug that mimics a cannabinoid receptor ligand, the researchers found. Ligands are molecules that function by binding to specific receptors.
This therapy may help the cancer research team to found out the caner in early stage.
1. www.washingtonpost.com/wp-dyn/content/article/2008/08/01/AR2008080100937.html
2. www.medicinenet.com/script/main/art.asp?articlekey=91511
3. hightimes.com/news/dan/4542
copy paste link
4. neurotalk.psychcentral.com/thread51199.html
 |
|
|---|---|
 |
|
| (−)-(6aR,10aR)-6,6,9-trimethyl- 3-pentyl-6a,7,8,10a-tetrahydro- 6H-benzo[c]chromen-1-ol |
|
Tetrahydrocannabinol (THC), or more precisely its main isomer (−)-trans-Δ9-tetrahydrocannabinol ((6aR,10aR)-delta-9-tetrahydrocannabinol), is the principal psychoactive constituent (or cannabinoid) of the cannabis plant. First isolated in 1964, in its pure form, by Israeli scientists Raphael Mechoulam, Yechiel Gaoni and colleagues at the Hebrew University of Jerusalem, it is a glassy solid when cold, and becomes viscous and sticky if warmed. A pharmaceutical formulation of (−)-trans-Δ9-tetrahydrocannabinol, known by its INN dronabinol, is available by prescription in the U.S. and Canada under the brand name Marinol. An aromatic terpenoid, THC has a very low solubility in water, but good solubility in most organic solvents, specifically lipids and alcohols.
Like most pharmacologically-active secondary metabolites of plants, THC in cannabis is assumed to be involved in self-defense, perhaps against herbivores. THC also possesses high UV-B (280–315 nm) absorption properties, which, it has been speculated, could protect the plant from harmful UV radiation exposure.
Tetrahydrocannabinol with double bond isomers and their stereoisomers is one of only three cannabinoids scheduled by Convention on Psychotropic Substances (the other two are dimethylheptylpyran and parahexyl). Note that cannabis as a plant is scheduled by Single Convention on Narcotic Drugs (Schedule I and IV).
Dr. Reddy’s Announces the Launch of Decitabine for Injection
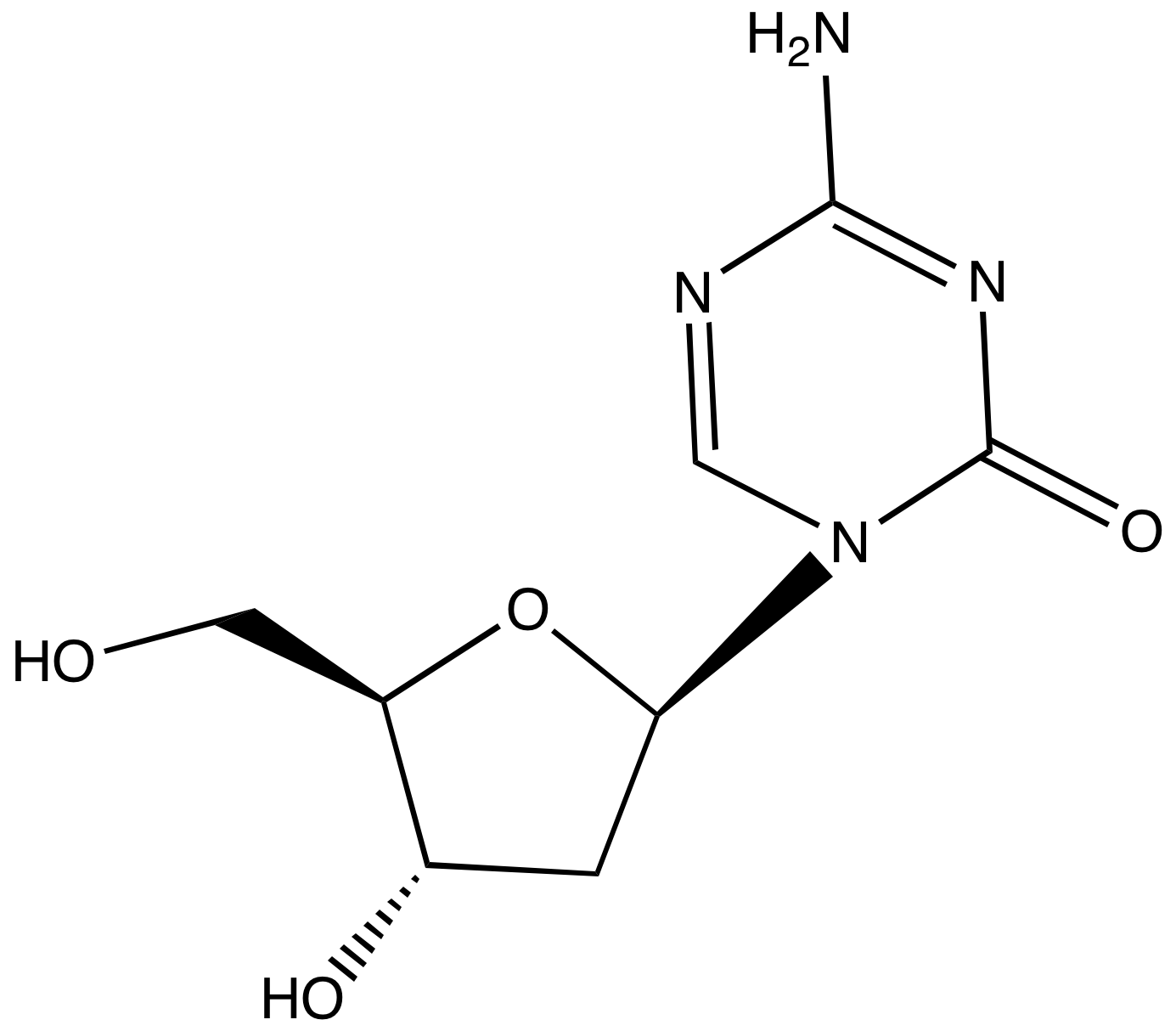
Decitabine
Hyderabad, India, July 12, 2013 — Dr. Reddy’s Laboratories announced today that it has launched Decitabine for Injection (50mg) a therapeutic equivalent generic version of Dacogen (Decitabine for Injection) in the US market on July 11, 2013, following the approval by the United States Food & Drug Administration (USFDA) of Dr. Reddy’s ANDA for Decitabine for Injection.
The Dacogen brand has U.S. sales of approximately $260 Million MAT for the most recent twelve months ending in July 2013 according to IMS Health*.
Dr. Reddy’s Decitabine for Injection 50 mg is available as a single dose vial.
About Dr. Reddy’s

Dr. Reddy’s Laboratories Ltd. (NYSE: RDY) is an integrated global pharmaceutical company, committed to providing affordable and innovative medicines for healthier lives. Through its three businesses – Pharmaceutical Services and Active Ingredients, Global Generics and Proprietary Products – Dr. Reddy’s offers a portfolio of products and services including APIs, custom pharmaceutical services, generics, biosimilars, differentiated formulations and NCEs. Therapeutic focus is on gastro-intestinal, cardiovascular, diabetology, oncology, pain management, anti-infective and pediatrics. Major markets include India, USA, Russia and CIS, Germany, UK, Venezuela, S. Africa, Romania, and New Zealand. For more information, log on to: http://www.drreddys.com.
Dacogen® is a registered trademark used by Eisai Inc. under license from Astex Pharmaceuticals, Inc
Decitabine (trade name Dacogen), or 5-aza-2′-deoxycytidine, is a drug for the treatment of myelodysplastic syndromes, a class of conditions where certain blood cells are dysfunctional, and for acute myeloid leukemia (AML).[1] Chemically, it is a cytidine analog.
Decitabine is a hypomethylating agent.[2][3] It hypomethylates DNA by inhibiting DNA methyltransferase.
It functions in a similar manner to azacitidine, although decitabine can only be incorporated into DNA strands while azacitidine can be incorporated into both DNA and RNA chains.

Clinical uses
Decitabine is indicated for the treatment of myelodysplastic syndromes (MDS) including previously treated and untreated, de novo and secondary MDS of all French-American-British subtypes (refractory anemia, refractory anemia with ringed sideroblasts, refractory anemia with excess blasts, refractory anemia with excess blasts in transformation, and chronic myelomonocytic leukemia) and Intermediate-1, Intermediate-2, and High-Risk International Prognostic Scoring System groups. In patients with renal insufficiency, Batty and colleagues reported the first case series on the feasibility of therapy with hypomethylating agents in patients with renal insufficiency.[4]
Chemical synthesis
Decitabine can be synthesized from a benzoyl-protected chlorosugar:[5] 
- “EC Approves Marketing Authorization Of DACOGEN For Acute Myeloid Leukemia”. 2012-09-28. Retrieved 28 September 2012.
- Kantarjian H, Issa JP, Rosenfeld CS, et al. (April 2006). “Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study”. Cancer 106 (8): 1794–803. doi:10.1002/cncr.21792. PMID 16532500.
- Kantarjian HM, O’Brien S, Cortes J, et al. (August 2003). “Results of decitabine (5-aza-2’deoxycytidine) therapy in 130 patients with chronic myelogenous leukemia”. Cancer 98 (3): 522–8. doi:10.1002/cncr.11543. PMID 12879469.
- Ravandi, F.; Cortés, J. E.; O’Brien, S.; Pierce, S.; Garcia-Manero, G.; McCue, D.; Santos, F. P. S.; Jabbour, E. et al. (2010). “Feasibility of Therapy with Hypomethylating Agents in Patients with Renal Insufficiency”. Clinical Lymphoma, Myeloma & Leukemia 10 (3): 205–210. doi:10.3816/CLML.2010.n.032. PMID 20511166.
|displayauthors=suggested (help) edit - Piml, J.; Sorm, F. (1964). Coll. Czech. Chem. Commun. 29: 2576.

FDA Approves New Drug Gilotrif for Advanced Lung Cancer

AFATINIB
FRIDAY July 12, 2013 — A new drug to treat advanced lung cancer has been approved by the U.S. Food and Drug Administration.
Gilotrif (afatinib) is approved to treat patients with a specific subtype of of non-small cell lung cancer (NSCLC). About 85 percent of lung cancers are NSCLC, making it the most common type of lung cancer.
Gilotrif is approved to treat tumors that carry key deletions on the epidermal growth factor receptor (EGFR) gene, long a target for lung cancer therapeutics. Mutations in the EGFR gene are thought to occur in about 10 percent of non-small cell lung cancers, and most of those mutations are targeted by Gilotrif, the FDA said.http://www.drugs.com/news/fda-approves-new-gilotrif-advanced-lung-cancer-45917.html
Afatinib (INN; planned trade name Tomtovok, previously Tovok) is a candidate drug against non-small cell lung carcinoma (NSCLC), developed by Boehringer Ingelheim. As of July 2012, it is undergoing Phase III clinical trials for this indication and breast cancer, as well as Phase II trials for prostateand head and neck cancer,and a Phase I glioma trial , Afatinib is not a first-line treatment; it is only used after other therapies have failed.
In October 2010 a Phase III trial in NSCLC patients called Lux-Lung 5 began with this drug Fall 2010 interim results suggested the drug extended progression-free survival threefold compared to placebo, but did not extend overall survival.In May 2012, the Phase IIb/III trial Lux-Lung 1 came to the same conclusion.
Phase II results for breast cancer that over-expresses the protein human epidermal growth factor receptor 2 (Her2-positive breast cancer) were described as promising by the authors, with 19 of 41 patients achieving benefit from afatinib. Double-blind Phase III trials are under way to confirm or refute this finding. Her2-negative breast cancers showed limited or no response to the drug
FDA Accepts the Filing of The Medicines Company’s New Drug Application for Intravenous Antiplatelet Agent Cangrelor
We know that, Cangrelor is a P2Y12 inhibitor under investigation as an antiplatelet drug for intravenous application. Some P2Y12 inhibitors are used clinically as effective inhibitors of adenosine diphosphate-mediated platelet activation and aggregation. Unlike clopidogrel (Plavix), which is a prodrug, cangrelor is an active drug not requiring metabolic conversion. Now…
