
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Morphine and oxycodone Dual-Opioid combination (MoxDuo)
SYDNEY and BEDMINSTER, N.J., March 14, 2013
QRxPharma Limited announced today the US Food and Drug Administration (FDA) has set 26 August 2013 as the Prescription Drug User Fee Act (PDUFA) date for action on the Company’s resubmitted MoxDuo New Drug Application (NDA).
“We are pleased that the FDA has formally accepted our resubmitted MoxDuo NDA” said Dr. John Holaday , Managing Director and Chief Executive Officer, QRxPharma. “We expect the Advisory Committee meeting to be scheduled between late June and late July and will update shareholders once formal notification has been received,” added Holaday.
The NDA is the basis for recommencing the regulatory approval process for MoxDuo for the treatment of moderate to severe acute pain, a $2.5 billion segment of the $8 billion spent annually on prescription opioids in the US. MoxDuo, an immediate release Dual Opioid® pain therapy, is a patented 3:2 fixed ratio combination of morphine

and
oxycodone

FDA Sets 26 August 2013 As New PDUFA Date For MoxDuo NDA.
New Drugs May Offer Hope to Parkinson’s Patients
The research findings will be presented at the annual meeting of the American Academy of Neurology in San Diego from March 16 to 23 2013
THURSDAY March 14, 2013 — Parkinson’s disease has no cure, but three experimental treatments may help patients cope with unpleasant symptoms and related problems, according to new research.
The research findings will be presented at the annual meeting of the American Academy of Neurology in San Diego from March 16 to 23.
“Progress is being made to expand our use of medications, develop new medications and to treat symptoms that either we haven’t been able to treat effectively or we didn’t realize were problems for patients,” said Dr. Robert Hauser, professor of neurology and director of the University of South Florida Parkinson’s Disease and Movement Disorders Center in Tampa.
Parkinson’s disease, a degenerative brain disorder, affects more than 1 million Americans. It destroys nerve cells in the brain that make dopamine, which helps control muscle movement. Patients experience shaking or tremors, slowness of movement, balance problems and a stiffness or rigidity in arms and legs.
In one study, Hauser evaluated the drug droxidopa, which is not yet approved for use in the United States, to help patients who experience a rapid fall in blood pressure when they stand up, which causes light-headedness and dizziness. About one-fifth of Parkinson’s patients have this problem, which is due to a failure of the autonomic nervous system to release enough of the hormone norepinephrine when posture changes.
Hauser studied 225 people with this blood-pressure problem, assigning half to a placebo group and half to take droxidopa for 10 weeks. The drug changes into norepinephrine in the body.
Those on the medicine had a two-fold decline in dizziness and lightheadedness compared to the placebo group. They had fewer falls, too, although it was not a statistically significant decline.
In a second study, Hauser assessed 420 patients who experienced a daily “wearing off” of the Parkinson’s medicine levodopa, during which their symptoms didn’t respond to the drug. He compared those who took different doses of a new drug called tozadenant, which is not yet approved, with those who took a placebo. All still took the levodopa.
At the start of the study, the patients had an average of six hours of “off time” a day when symptoms reappeared. After 12 weeks, those on a 120-milligram or 180-milligram dose of tozadenant had about an hour less of “off time” each day than they had at the start of the study.
Tozadenant, which works on brain receptors thought to regulate motor function, merits further study in future trials, Hauser said.

In another study, Hauser looked at 321 patients with early stage Parkinson’s whose symptoms weren’t handled well by a medicine called a dopamine agonist, typically the first drug prescribed for Parkinson’s patients. During the 18-week study, Hauser assigned them to take either their usual medicine plus an add-on drug called rasagiline (brand name Azilect) or their usual medicine and a placebo.\

Azilect is approved for use in patients with early stage disease as a single therapy or as an add-on to levodopa, Hauser said, but not yet as an add-on to dopamine agonists.
Those taking the Azilect — but not those taking the placebo — improved by 2.4 points on a standard Parkinson’s disease rating scale.
Costs of the still unapproved drugs are not known. Azilect costs about $200 monthly at the 1-milligram daily dose used in the study.
Each of the studies was funded by the pharmaceutical company making the particular drug: Chelsea Therapeutics paid for the blood-pressure study; Biotie Therapies Inc., supported the “wearing-off” study; and Teva Pharmaceutical Industries sponsored the Azilect study. Hauser is a consultant for all three companies.
Most impressive of the three studies is the use of droxidopa to prevent dizziness and fainting, said Dr. Michael Okun, national medical director of the National Parkinson Foundation and director of the University of Florida Center for Movement Disorders and Neurorestoration.

Drugs are already available to treat the problem, and compression stockings are also often recommended. Even so, “having another drug in that arena is going to help a lot of people,” he said.
The effects of the other two treatments are more modest, said Okun, who is also a neurology professor. Additional studies will help determine how noteworthy the effects are in real life, he said.
Findings presented at medical meetings should be considered preliminary until published in a peer-reviewed medical journal.
More information
To learn more about Parkinson’s disease, visit the National Parkinson Foundation.
Sinovac Reports Preliminary Top-Line Results from Phase III Clinical Trial for EV71 Vaccine Candidate Against Hand, Foot and Mouth Disease
")
BEIJING, March 14, 2013
Sinovac Biotech Ltd. a leading provider of vaccines in China, announced today preliminary top-line data from its Phase III clinical trial assessing the efficacy, immunogenicity and safety of the Company’s proprietary Enterovirus 71 (“EV71”) vaccine against hand, foot and mouth disease (“HFMD”).
The primary objective of the study was to evaluate the efficacy of the EV71 vaccine in the prevention of HFMD caused by EV71 in infants of 6 to 35 months old. The preliminary Phase III data showed that Sinovac’s EV71 vaccine was 95.4% (95% CI: 87.5%, 98.3%) efficacious against HFMD caused by EV71.
The Phase III trial showed good immunogenicity and safety for Sinovac’s EV71 vaccine. The overall incidence of serious adverse events in this trial was 2.2% among the EV71 candidate vaccine recipients and 2.6% among those receiving a control vaccine during the fourteen months observation period. The difference in rates of serious adverse events (“SAEs”) is not statistically significant. Most of the SAEs were considered unlikely to be vaccine-related.
The double-blinded, randomized, placebo controlled Phase III clinical trial was conducted at three sites across China’s Jiangsu province. Approximately 10,000 healthy infants completed the two dose vaccination schedule (at 0 and 28 days) in the first quarter of 2012, prior to the HFMD epidemic season in China, followed by active monitoring period.
In parallel, Sinovac conducted another clinical study that was comprised of 1,400 volunteers and designed to evaluate the consistency of three consecutive lots of EV71 vaccine manufactured by the Company. The trial was conducted in children from 6 month to 5 years old. After receiving the vaccine, the ratios of neutralizing antibody GMTs on the 56th day of any two groups were calculated and the 95% confidence intervals of the ratios are all between 0.67 and1.5, which indicates the immunogenicity of the three vaccine lots is equivalent. The study results showed consistent immune response for all three lots and a good safety profile. With immunogenicity equivalent across the three consecutive lots, the results showed Sinovac’s vaccine production process and quality are stable.
In March 2008, an EV71 outbreak in Fuyang City of China’s Anhui Province caused 23 fatalities, and attracted significant attention from the government and medical communities. In May 2008, the PRC Ministry of Health identified EV71 as a Class C infectious disease according to prevention and control regulations. EV71 outbreaks have increased over the last five years, with over 1 million cases identified and 500 to 900 reported fatalities each year.
Dr. Weidong Yin , Chairman, President and CEO of Sinovac, commented, “We are excited to report an over 95% efficacy rate from the Phase III trial on our proprietary EV71 vaccine candidate. The conclusion of this trial marks an important milestone in the development of our proprietary vaccine. Hand, foot, and mouth disease continues to represent a significant unmet public health need and economic burden in China, as well as several other Asian countries. Our EV71 vaccine is poised to provide an effective solution to prevent hand, food and mouth disease caused by EV71, a much needed resource given the current limited prevention and EV71 specific treatment methods. At Sinovac, we are committed to our stated mission to develop and supply vaccines to eliminate human diseases.”
Professor Hua Wang, Lead Principal Investigator, stated, “The Phase III study for Sinovac’s EV71 vaccine candidate met its primary objective. The trial results demonstrated that the vaccine is not only safe, but shows significant efficacy in subjects.”
The Company’s next step is to finalize the clinical report, which will become an important part of documents to be filed with the PRC State Food and Drug Administration (“SFDA”) for the application of new drug certificate, GMP certification, and the production license in order to commence the commercial production of the vaccine. In parallel, Sinovac’s dedicated EV71 vaccine manufacturing facility has been completed and is ready for the GMP inspection by SFDA.
Sinovac obtained clinical research approval for its proprietary EV71 vaccine candidate from the SFDA in December 2010, and completed Phase I and II clinical trials in 2011. The preliminary results of the Phase I and Phase II studies confirmed that Sinovac’s vaccine candidate has good safety and immunogenicity profile.
About Sinovac
Sinovac Biotech Ltd. is a China-based biopharmaceutical company that focuses on research, development, manufacturing and commercialization of vaccines that protect against human infectious diseases including hepatitis A and B, seasonal influenza, H5N1 pandemic influenza and mumps, as well as animal rabies vaccine. In 2009, Sinovac was the first company worldwide to receive approval for its H1N1 influenza vaccine, Panflu.1, and has manufactured it for the Chinese Central Government, pursuant to the government-stockpiling program. The Company is also the only supplier of the H5N1 pandemic influenza vaccine to the government-stockpiling program. Sinovac is developing a number of new pipeline vaccines including vaccines for enterovirus 71 (against hand, foot, and mouth disease), pneumococcal conjugate, pneumococcal polysaccharides, varicella and rubella. Sinovac sells its vaccines mainly in China and exports selected vaccines to Mongolia, Nepal, and the Philippines.

A sensor-adaptor mechanism for enterovirus uncoating from structures of EV71
Xiangxi Wang, Wei Peng, Jingshan Ren, Zhongyu Hu, Jiwei Xu, Zhiyong Lou, Xumei Li, Weidong Yin, Xinliang Shen, Claudine Porta, Thomas S Walter, Gwyndaf Evans, Danny Axford, Robin Owen, David J Rowlands, Junzhi Wang*, David I Stuart*, Elizabeth E Fry* & Zihe Rao*
Enterovirus 71 1 (EV71) is a major agent of hand, foot and mouth disease in children that can cause severe central nervous system disease and death. No vaccine or antiviral therapy is available. High-resolution structural analysis of the mature virus and natural empty particles shows that the mature virus is structurally similar to other enteroviruses. In contrast, the empty particles are markedly expanded and resemble elusive enterovirus-uncoating intermediates not previously characterized in atomic detail. Hydrophobic pockets in the EV71 1 capsid are collapsed in this expanded particle, providing a detailed explanation of the mechanism for receptor-binding triggered virus uncoating. These structures provide a model for enterovirus uncoating in which the VP1 1 GH loop acts as an adaptor-sensor for cellular receptor attachment, converting heterologous inputs to a generic uncoating mechanism, highlighting new opportunities for therapeutic intervention. [ Nat Struct Mol Biol. 2012 Mar 4. doi: 10.1038/nsmb.2255. Epub ahead of print. PMID: 22388738 ][ PDF ]
Biogen submits haemophilia A drug to FDA

Mar 14 2013
Biogen Idec has filed the first long-lasting Factor VIII treatment for haemophilia A with the US Food and Drug Administration.
The US biotech major has submitted recombinant factor VIII Fc fusion protein (rFVIIIFc), the first haemophilia A product candidate “in a new class of long-lasting clotting factor therapies being developed with the goal of reducing the burden of treatment for this condition”. If approved, rFVIIIFc will be the first major advance in haemophilia A treatment in more than two decades, Biogen claims.
The regulatory submission is based on results from A-LONG, the largest Phase III study in haemophilia A to date. Glenn Pierce, Biogen’s head of global medical affairs, noted that in that trial, patients were able to inject rFVIIIFc once-weekly to twice-weekly, “which creates the potential for those currently on prophylactic treatment to reduce injections by 50 to 100 per year”. Moreover, patients currently treating bleeding episodes could potentially dose once per week “and maintain significant protection from bleeding with about the same total number of injections each year they use to treat bleeding episodes today”, he added.
Earlier this month, the FDA accepted for review the company’s BLA for its factor IX candidate, rFIXFc, for use in patients with haemophilia B.
Links
Boehringer Ingelheim Announces Interim Results Evaluating Virologic Response Rates in HCV/HIV Co-Infected Patients Treated with Faldaprevir
Ridgefield, CT, March 4, 2013 – Today Boehringer Ingelheim Pharmaceuticals, Inc. announced the first interim results in HCV/HIV co-infected patients from the company’s ongoing hepatitis C (HCV) clinical trial program, HCVersoTM. These results, from the Phase 3 trial STARTVersoTM 4, were presented today at the 20th annual Conference on Retroviruses and Opportunistic Infections (CROI) in Atlanta, GA.

The interim results showed that 80% of HCV/HIV co-infected patients achieved early treatment success (ETS)*, as defined by the study protocol, when given an investigational HCV regimen that included faldaprevir (BI 201335). Results were consistent across patients regardless of HIV therapy or prior HCV treatment status, including patients who were HCV treatment-naïve or had previously relapsed during HCV treatment with pegylated interferon and ribavirin (PegIFN/RBV). Patients who achieved ETS were eligible for randomization to a…
View original post 688 more words
FDA Approves Lymphoseek
FDA Approves Lymphoseek to Help Locate Lymph Nodes in Patients with Certain Cancers
March 13, 2013 — The U.S. Food and Drug Administration today approved Lymphoseek (technetium Tc 99m tilmanocept) Injection, a radioactive diagnostic imaging agent that helps doctors locate lymph nodes in patients with breast cancer or melanoma who are undergoing surgery to remove tumor-draining lymph nodes.
Lymph nodes filter lymphatic fluid that flows from the body’s tissues. This fluid may contain cancer cells, especially if the fluid drains a part of the body containing a tumor. By surgically removing and examining the lymph nodes that drain a tumor, doctors can sometimes determine if a cancer has spread.
Lymphoseek is an imaging drug that helps locate lymph nodes; it is not a cancer imaging drug. Lymphoseek is the first new drug used for lymph node mapping to be approved in more than 30 years. Other FDA-approved…
View original post 200 more words
DRUG REVIEW- Tigecycline

Tigecycline
N-[(5aR,6aS,7S,9Z,10aS)-9-[amino(hydroxy)methylidene]-4,7-bis(dimethylamino)-1,10a,12-trihydroxy-8,10,11-trioxo-5,5a,6,6a,7,8,9,10,10a,11-decahydrotetracen-2-yl]-2-(tert-butylamino)acetamide
Tigecycline is a glycylcycline antibiotic[1][2] developed by Francis Tally[3] and marketed by Wyeth under the brand name Tygacil. It was given a U.S. Food and Drug Administration (FDA) fast-track approval and was approved on June 17, 2005. It was developed in response to the growing prevalence of antibiotic resistance in bacteria such as Staphylococcus aureus and Acinetobacter baumannii. The New Delhi metallo-β-Lactamase multidrug-resistant Enterobacteriaceae has also shown susceptibility to tigecycline.[4]
This antibiotic is the first clinically available drug in a new class of antibiotics called the glycylcyclines. It is structurally similar to the tetracyclines in that it contains a central four-ring carbocyclic skeleton and is actually a derivative of minocycline. Tigecycline has a substitution at the D-9 position which is believed to confer broad spectrum activity.
Tigecycline is bacteriostatic and is a protein synthesis inhibitor by binding to the 30S ribosomal subunit of bacteria and thereby blocking entry of Aminoacyl-tRNA into the A site of the ribosome during prokaryotic translation.[5]
Tigecycline is given intravenously and has activity against a variety of gram-positive and gram-negative bacterial pathogens, many of which are resistant to existing antibiotics. Tigecycline successfully completed phase III trials in which it was at least equal to intravenous vancomycin and aztreonam to treat complicated skin and skin structure infections (cSSSI), and to intravenous imipenem and cilastatin to treat complicated intra-abdominal infections (cIAI).[6] Tigecycline is active against many Gram-positive bacteria, Gram-negative bacteria and anaerobes – including activity against methicillin-resistant Staphylococcus aureus (MRSA), Stenotrophomonas maltophilia, Haemophilus influenzae, and Neisseria gonorrhoeae (with MIC values reported at 2mcg/mL) and multi-drug resistant strains of Acinetobacter baumannii. It has no activity against Pseudomonas spp. or Proteus spp. The drug is licenced for the treatment of skin and soft tissue infections as well as intra-abdominal infections.
Tigecycline is given by slow intravenous infusion (30 to 60 minutes). A single dose of 100 mg is given first, followed by 50 mg every twelve hours after that. Patients with impaired liver function need to be given a lower dose. No adjustment is needed for patients with impaired kidney function. It is not licensed for use in children. There is no oral form available.
Tigecycline has similar side effects to the tetracyclines. The most common side effects of tigecycline are diarrhea, nausea and vomiting. Nausea and vomiting is mild or moderate and usually occurs during the first two days of therapy. Other side effects include pain at the injection site, swelling and irritation; increased or decreased heart rate and infections. Also avoid use in children and pregnancy, due to its effects on teeth and bone. As with other antibiotics, overgrowth of organisms that are not susceptible to tigecycline can occur.
Tigecycline showed an increased mortality in patients treated for hospital-acquired pneumonia, especially ventilator-associated pneumonia, but also in patients with complicated skin and skin structure infections, complicated intra-abdominal infections and diabetic foot infection.[7]
It may have potential for use in acute myeloid leukaemia.[8]
- GAR-936[9]
- Tygacil
References
- Rose W, Rybak M (2006). “Tigecycline: first of a new class of antimicrobial agents.”. Pharmacotherapy 26 (8): 1099–110. doi:10.1592/phco.26.8.1099. PMID 16863487.
- Kasbekar N (2006). “Tigecycline: a new glycylcycline antimicrobial agent.”. Am J Health Syst Pharm 63 (13): 1235–43. doi:10.2146/ajhp050487. PMID 16790575.
- Projan, Steven J (Jan. 2010). “Francis Tally and the discovery and development of tigecycline: a personal reminiscence”. Clin. Infect. Dis. (United States) 50 Suppl 1: S24–5. doi:10.1086/647941. PMID 20067389.
- Kumarasamy et. al.; Toleman, Mark A; Walsh, Timothy R; Bagaria, Jay; Butt, Fafhana; Balakrishnan, Ravikumar; Chaudhary, Uma; Doumith, Michel et al. (2010). “Emergence of a new antibiotic resistance mechanism in India, Pakistan, and the UK: a molecular, biological, and epidemiological study”. The Lancet Infectious Diseases 10 (9): 597–602. doi:10.1016/S1473-3099(10)70143-2. PMC 2933358. PMID 20705517.
- Tigecycline: A Novel Broad-Spectrum Antimicrobial: Pharmacology and Mechanism of Action Christine M. Slover, PharmD, Infectious Diseases Fellow, Keith A. Rodvold, PharmD and Larry H. Danziger, PharmD, Professor, Department of Pharmacy Practice, University of Illinois at Chicago, Chicago, IL
- Scheinfeld N (2005). “Tigecycline: a review of a new glycylcycline antibiotic.”. Journal of Dermatological Treatment 16 (4): 207–12. doi:10.1080/09546630510011810. PMID 16249141.
- http://www.fda.gov/Drugs/DrugSafety/ucm224370.htm
- Skrtić M, Sriskanthadevan S, Jhas B, Gebbia M, Wang X, Wang Z, Hurren R, Jitkova Y, Gronda M, Maclean N, Lai CK, Eberhard Y, Bartoszko J, Spagnuolo P, Rutledge AC, Datti A, Ketela T, Moffat J, Robinson BH, Cameron JH, Wrana J, Eaves CJ, Minden MD, Wang JC, Dick JE, Humphries K, Nislow C, Giaever G, Schimmer AD (2011) Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell 20(5):674-688
- Betriu C, Rodríguez-Avial I, Sánchez BA, Gómez M, Picazo JJ (2002). “Comparative in vitro activities of tigecycline (GAR-936) and other antimicrobial agents against Stenotrophomonas maltophilia“. J Antimicrob Chemother 50 (5): 758–59. doi:10.1093/jac/dkf196. PMID 12407139.
EMA
Human medicines European Public Assessment Report EPAR : Tygacil, tigecycline, Revision: 16, Authorised
http://www.ema.europa.eu/docs/en_GB/document_library/Referrals_document/Cetero_31/WC500136265.pdf
Phase III trial of Merck’s Vytorin passes vital safety test
mar 13 2013
Merck & Co’s stock enjoyed a boost yesterday after it revealed it has been given permission to continue a late-stage trial of its cholesterol buster Vytorin.
The Whitehouse Station, New Jersey-based firm must have a breathed a sigh of relief when the Data Safety Monitoring Board issued a green light for the Phase III IMPROVE for a second time, having found no significant safety concerns raised by the data.
After an earlier planned review of data last year, the Board, rather unusually, said it would undertake a second interim analysis at a later date, which had led to some concerns that there may be issues that could lead to the trial being halted, according to media reports.
However, it seems these fears are unfounded at this point, as the18,000-plus patient study – which is designed to determine whether Vytorin is more effective at reducing the risk of heart attack, stroke and death in patients with heart disease than simvastatin alone – has been cleared to conclude.
The drugmaker said the trial should finish in September next year, and it will no doubt be hoping for a positive outcome to prove the benefits of Vytorin – a combination of the generic simvastatin and the still-patented Zetia (ezetimibe) – and breathe a little new life into its heart franchise.
Citi Investment Research analyst Andrew Baum, however, expressed doubt in a research note the final analysis will show Merck’s drug is more effective than generic competition, according to the Associated Press.
 |
|
|---|---|
 |
|
| Combination of | |
| Ezetimibe | via Niemann-Pick C1-Like 1 protein |
| Simvastatin | Statin HMG-CoA reductase inhibitor |
Cangrelor, AR-C69931MX Shows Improvement Over Plavix in Phase III Trial

Cangrelor, AR-C69931MX
[dichloro-[[[(2R,3S,4R,5R)-3,4-dihydroxy-5-[6-(2-methylsulfanylethylamino)-2-(3,3,3-trifluoropropylsulfanyl)purin-9-yl]oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-hydroxyphosphoryl]methyl]phosphonic acid
N-[2-(Methylthio)ethyl]-2-[(3,3,3-trifluoropropyl)thio]-5¢-adenylic acid monoanhydride with (dichloromethylene)bis[phosphonic acid]
163706-06-7 cas no
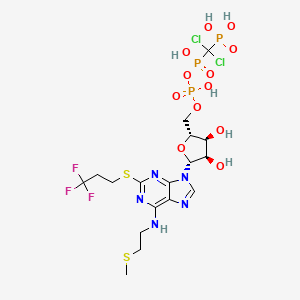 Cangrelor
CangrelorUPDATE
Company:
Approval Status:
Approved June 2015
Specific Treatments:
For reducing periprocedural thrombotic events
Therapeutic Areas
Company:
Approval Status:
Approved June 2015
Specific Treatments:
For reducing periprocedural thrombotic events
Therapeutic Areas
Kengreal (cangrelor)
MAR 09, 2013
The Medicines Company said yesterday it will pursue marketing approvals for its anti-clotting drug candidate Cangrelor after it met its primary efficacy endpoint in a Phase III clinical trial of improvement compared with Plavix (clopidogrel).
The intravenous small molecule antiplatelet agent reduced by 22% the likelihood of patients experiencing death, myocardial infarction, ischemia-driven revascularization, or stent thrombosis within 48 hours of taking it—to 4.7% from 5.9% of subjects randomized during CHAMPION PHOENIX. The Phase III trial compared Cangrelor to oral Plavix in 11,145 patients undergoing percutaneous coronary intervention.
Cangrelor also showed a 38% reduction (0.8% compared with 1.4%) over Plavix in the likelihood of patients experiencing the key secondary endpoint, incidence of stent thrombosis at 48 hours.
Cangrelor is designed to prevent platelet activation and aggregation that leads to thrombosis in acute care settings, including in patients undergoing percutaneous coronary intervention. During CHAMPION PHOENIX, Cangrelor made its best showing in patients with Q-wave myocardial infarction (QMI), lowering by 39% (to 0.2% compared with 0.3%) the incidence of QMI. Cangelor’s most disappoint showing was its inability to lower the odds of death compared with Clopidogrel; both drugs showed a likelihood of 0.3%.
“Our next step is to submit for market approvals in the U.S. and Europe. We anticipate submitting these data for a new drug application to the U.S. Food and Drug Administration in the second quarter with findings of prior trials, including the BRIDGE trial in patients awaiting open heart surgery,” Simona Skerjanec, PharmD, senior vp and innovation leader for antiplatelet therapies at The Medicines Company, said in a statement.
Cangrelor is a P2Y12 inhibitor under investigation as an antiplatelet drug[1] for intravenous application. Some P2Y12 inhibitors are used clinically as effective inhibitors of adenosine diphosphate-mediated platelet activation and aggregation.[1] Unlike clopidogrel (Plavix), which is a prodrug, cangrelor is an active drug not requiring metabolic conversion.
Poor interim results led to the abandonment of the two CHAMPION clinical trials in mid 2009.[2] The BRIDGE study, for short term use prior to surgery, continues.[3] The CHAMPION PHOENIX trial was a randomized study of over 11,000 patients published in 2013. It found usefulness of cangrelor in patients getting cardiac stents. Compared with clopidogrel given around the time of stenting, intravenous ADP-receptor blockade with cangrelor significantly reduced the rate of stent thrombosis and myocardial infarction.[4] Reviewers have questioned the methodology of the trial.[5]

One particularly preferred example of a reversible, short-acting P2Y12 inhibitor is cangrelor. Cangrelor is a potent, direct, and reversible antagonist of the platelet P2Y12 receptor. Cangrelor has a half-life of approximately less than 10 minutes, allowing for a return to normal platelet function in a very short period of time upon discontinuation of the drug. By reducing the need for a compound to be metabolized for activity, and by having a relatively short half-life, reversible, short-acting P2Y12 inhibitors are considered “reversible,” meaning that full platelet functionality may return rather quickly as compared to thienopyridines.
The binding of cangrelor to the P2Y12 receptor inhibits platelet activation as well as aggregation when mediated in whole or in part via this receptor. Cangrelor can be derived completely from synthetic materials, and is an analogue of adenosine triphosphate (ATP). ATP is a natural antagonist of the P2Y12 receptor sites and is found in humans.
The chemical structure for cangrelor is depicted below as Formula I.

Cangrelor is clinically well tolerated and safe and has no drug-drug interaction with aspirin, heparin or nitroglycerin. Unlike orally dosed thienopyridines, cangrelor can be administered intravenously and binds directly to P2Y12 receptor sites of platelets. In each of the embodiments of the present invention, the term “cangrelor” encompasses the compound of Formula I as well as tautomeric, enantiomeric and diastereomeric forms thereof, and racemic mixtures thereof, other chemically active forms thereof, and pharmaceutically acceptable salts of these compounds, including a tetrasodium salt. These alternative forms and salts, processes for their production, and pharmaceutical compositions comprising them, are well known in the art and set forth, for example, in U.S. Pat. No. 5,721,219. Additional disclosure relevant to the production and use of cangrelor may be found in U.S. Pat. Nos. 5,955,447, 6,130,208 and 6,114,313, as well as in U.S. Appln. Publication Nos. 2006/0270607 and 2011/0112030.
Invasive procedures means any technique where entry to a body cavity is required or where the normal function of the body is in some way interrupted by a medical procedure and/or treatment that invades (enters) the body, usually by cutting or puncturing the skin and/or by inserting instruments into the body. Invasive procedures can include coronary artery bypass grafting (CABG), orthopedic surgeries, urological surgeries, percutaneous coronary intervention (PCI), other general invasive procedures, such as endarterectomy, renal dialysis, cardio-pulmonary bypass, endoscopic procedures or any medical, surgical, or dental procedure that could result in excessive bleeding or hemorrhage to the patient.
Perioperative means the period of a patient’s invasive procedure which can occur in hospitals, surgical centers or health care providers’ offices. Perioperative includes admission, anesthesia, surgery, to recovery.
Thrombosis is the formation of a blood clot (thrombus) inside a blood vessel obstructing the flow of blood through the circulatory system. When a blood vessel is injured, the body uses platelets and fibrin to form a blood clot to prevent blood loss. Some examples of the types of thrombosis include venous thrombosis which includes deep vein thrombosis, portal vein thrombosis, renal vein thrombosis, jugular vein thrombosis, Budd-Chiari syndrome, Paget-Schroetter disease, cerebral venous sinus thrombosis, cerebral venous sinus thrombosis and arterial thrombosis which includes stroke and myocardial infarction.
The compound cangrelor from the Medicines Company is represented by the structure

 OR
ORRN: 163706-36-3



…………
J. Med. Chem., 1999, 42 (2), pp 213–220
http://pubs.acs.org/doi/full/10.1021/jm981072s
10l (AR-C69931MX)
N6–(2-Methylthioethyl)-2-(3,3,3-trifluoropropylthio)-5‘-adenylic Acid, Monoanhydride withDichloromethylenebis(phosphonic acid) (10l). Prepared as the triammonium salt in 4% yield from 3l: 1H NMR δ(D2O) 8.30 (1H, s, H8), 5.97 (1H, d, J = 5.5 Hz, H1‘), 4.65 (1H, m, H2‘), 4.47 (1H, m, H3‘), 4.28 (1H, m, H4‘), 4.17 (2H, m, H5‘a and H5‘b), 3.67 (br s, NHCH2), 3.21 (2H, t, J = 7.6 Hz, SCH2), 2.72 (2H, t, J = 6.6 Hz, SCH2CH2CF3), 2.58 (2H, m, NCH2CH2), 2.04 (3H, s, SCH3);31P NMR δ(D2O) 8.80 (d, 1P, J = 18.6 Hz, Pγ), 0.42 (dd, 1P, J1 = 18.9 Hz, J2 = 28.9 Hz, Pβ), −9.41 (d, 1P, J = 29.0 Hz, Pα). Anal. (C17H34Cl2F3N8O12P3S2·3H2O) H, N, S; C: calcd, 23.16; found, 23.66.
References
- Cangrelor Attenuates Coated-Platelet Formation
- CHAMPION Trials With Cangrelor Stopped for Lack of Efficacy
- What Cangrelor Failure Means to Medicines
- Effect of Platelet Inhibition with Cangrelor during PCI on Ischemic Events (2013) Bhatt, DL etal. New England Journal of Medicine March 10, 2013 DOI: 10.1056/NEJMoa1300815 (published initially online).
- The Duel between Dual Antiplatelet Therapies (2013) Lange, RA and Hillis, LD. New England Journal of Medicine March 10, 2013 DOI: 10.1056/NEJMe1302504
- 15th European Federation for Medicinal Chemistry International Symposium on Medicinal Chemistry (Sept 6 1998, Edinburgh)1998,:Abst P.281
- Specific P2Y12 purinoceptor antagonist; inhibits ADP-induced platelet aggregation. Prepn: A. H. Ingall et al., WO 9418216 (1994 to Fisons); eidem, US 5721219 (1998 to Astra); and in vivo antithrombotic activity: idem et al., J. Med. Chem. 42, 213 (1999).
- In vivo antithrombotic effects in canine arterial thrombosis: J. Huang et al., J. Pharmacol. Exp. Ther. 295, 492 (2000).
- Mechanism of action study: A. Ishii-Watabe et al., Biochem. Pharmacol. 59, 1345 (2000).
- Clinical safety assessment and evaluation in acute coronary syndromes: R. F. Storey et al., Thromb. Haemostasis 85, 401 (2001); in angina pectoris and non-Q-wave myocardial infarction: F. Jacobsson et al., Clin. Ther. 24, 752 (2002).
- Clinical pharmacodynamics compared with clopidogrel: R. F. Storey et al., Platelets 13, 407 (2002).
- Review of clinical development: S. C. Chattaraj, Curr. Opin. Invest. Drugs2, 250-255 (2001).
- WO2013/103567 A2,
- Journal of Medicinal Chemistry, 1999 , vol. 42, 2 p. 213 – 220
Lesinurad
Lesinurad
Acetic acid, 2-[[5-bromo-4-(4-cyclopropyl-1-naphthalenyl)-4H-1,2,4-triazol-3-yl]thio]-,
sodium salt (1:1)
Sodium 2-{[5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-
yl]sulfanyl}acetate
2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)acetic acid
MOLECULAR FORMULA C17H13BrN3NaO2S
MOLECULAR WEIGHT 426.3
http://clinicaltrials.gov/show/NCT01508702
http://www.ama-assn.org/resources/doc/usan/lesinurad.pdf
Ardea Biosciences, Inc.
- Lesinurad
- RDEA 594
- RDEA594
- UNII-09ERP08I3W
Gout phase 3
Gout is associated with elevated levels of uric acid that crystallize and deposit in joints, tendons, and surrounding tissues. Gout is marked by recurrent attacks of red, tender, hot, and/or swollen joints.
This study will assess the serum uric acid lowering effects and safety of lesinurad compared to placebo in patients who are intolerant or have a contraindication to allopurinol or febuxostat.
http://euroscan.org.uk/technologies/technology/view/2386
Lesinurad (RDEA-594, lesinurad sodium) is a selective urate transporter-1 (URAT-1) inhibitor, which blocks the reabsorption of urate within the renal proximal tubule. It is intended for the treatment of gout after failure of first line therapy and is administered orally at 400mg once daily
A Phase 3 Randomized, Double-Blind, Multicenter, Placebo- Controlled Study to Assess the Efficacy and Safety of Lesinurad Monotherapy Compared to Placebo in Subjects With Gout and an Intolerance or Contraindication to a Xanthine Oxidase Inhibitor
AstraZeneca’s lesinurad (formerly known as RDEA-594) is a selective oral Uric Acid Transporter URAT1 inhibitor currently in Phase III development for the treatment of of gout. The regulatory filings for lesinurad in the US and Europe are expected for the first half of 2014.

Gout (also known as podagra when it involves the big toe), while not life-threatening, is an excruciatingly painful condition caused by a buildup of a waste product in the blood called uric acid, which is normally eliminated from the body through urine. Excess Uric acid crystallizes and get deposited in the joints (usually the big toes), creating symptoms similar to an acute arthritis flare. Gout has seen a recent gradual resurgence as a result of rising obesity rates and poor diet according to a study in the journal Annals of the Rheumatic Diseases.
The current Standard treatment for gout works by inhibiting a protein called xanthine oxidase that helps in the formation of the uric acid. These therapies, some of which have been used for more than 50 years, are not effective in all patients. One is a generic drug called allopurinol that was approved in the U.S. in 1966. The other is febuxostat, marketed by Takeda Pharmaceutical Co. in the U.S. asUloric and by Ipsen SA and others in Europe as Adenuric and approved in the U.S. in 2009.
AstraZeneca’s new product Lesinurad, a selective uric acid re-absorption inhibitor (SURI), tackles gout by blocking a protein called Uric acid trasporter 1 (URAT1) that otherwise would cause the body to reabsorb the uric acid. AstraZeneca acquired lesinurad (aka RDEA-594) as part of its $1.26 billion takeouver of San Diego-based Ardea Biosciences in 2012. RDEA594 is a metabolite of RDEA806, a non-nucleoside reverse transcriptase inhibitor originally developed for HIV.

In top-line results from a Phase III LIGHT study released by AstraZeneca in December 2013 on gout patients who get no benefit from Zyloprim (allopurinol) and febuxostat, lesinurad alone significantly reduced serum levels of uric acid. The company has three other phase III studies ongoing that are testing the use of the drug alongside allopurinol and febuxostat, and these should generate results in the middle of 2014. Analysts at JPMorgan Chase forecast lesinurad alone may have peak sales of $1 billion a year. AstraZeneca also has a second, more potent drug called RDEA3179 to treat elevated levels of uric acid or hyperuricemia. Pfizer’s KUX-1151, licensed from Japan’s Kissei Phmarceuticals, is in early stage development.
Gout is not an automatic success indication of drugmakers. Savient Pharmaceuticals filed for Chapter 11 bankruptcy in October 2013 in the face of a severe cash crisis, having spent hundreds of millions of dollars on its would-be flagship — the gout-fighting drug Krystexxa (pegloticase) — with limited results. Krystexxa (pegloticase), a twice-monthly infusion designed to treat severe chronic gout that doesn’t respond to conventional therapy, was approved by the U.S. Food and Drug Administration in September 2010. Crealta Pharmaceuticals acquired Savient for $120.4 million in December 2013.
Lesinurad
RDEA-594
2-{[5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-yl]sulfanyl}acetic acid
CAS number: 878672-00-5 (Lesinurad), 1151516-14-1 (Lesinurad sodium)
Mechanism of Action:once-daily inhibitor of URAT1, a transporter in the kidney that regulates uric acid excretion from the body
US patents:US8242154 , US8173690, US808448
Indication: Gout
Developmental Status: Phase III (US, UK, EU)
Originator: Ardea Biosciences (Acquired by AstraZeneca for $1.26 billion in 2012)
Developer: AstraZeneca
…………………………
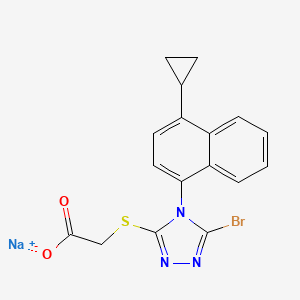
http://www.google.co.in/patents/US8242154
Example 8 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)acetic acid

Sodium hydroxide solution (2M aqueous, 33.7 mL, 67 mmol, 2 eq) was added to a suspension of 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)-N-(2-chloro-4-sulfamoylphenyl)acetamide (prepared by previously published procedures; 20 g, 34 mmol) in ethanol (200 mL) and the mixture heated at reflux for 4 hours. Charcoal (10 g) was added, the mixture stirred at room temperature for 12 hours and the charcoal removed by filtration. The charcoal was washed several times with ethanol and the filtrate then concentrated. Water (200 mL) was added and then concentrated to approx. one third volume, to remove all ethanol. Water (200 mL) and ethyl acetate (250 mL) were added, the mixture stirred vigorously for 15 mins and the organic layer removed. The aqueous layer was cooled to 0° C. and acidified by treatment with HCl (1N) resulting in the formation of a cloudy oily precipitate. The mixture was extracted with ethyl acetate (3×) and the combined organic extracts dried over sodium sulfate and concentrated to give 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)acetic acid as an off white solid (11.2 g, 82%).
Example 102 Methyl 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)acetate


Cyclopropylmagnesium bromide (150 mL, 0.5M in tetrahydrofuran) was slowly added to a solution of 1-bromonaphthalene (10 g, 50 mmol) and [1,3-bis(diphenylphosphino)propane]dichloro nickel (II) in tetrahydrofuran (10 mL) stirred at 0° C., and the reaction mixture stirred at room temperature for 16 hours. The solvent was removed under reduced pressure and ethyl acetate and aqueous ammonium chloride were added. After extraction, the organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield 1-cyclopropylnaphthalene (6.4 g, 76%).

Sodium nitrite (30 mL) was slowly added (over 2 hours) to 1-cyclopropylnaphthalene (6.4 g, 38 mmol) stirred at 0° C. The reaction mixture was stirred at 0° C. for an extra 30 min and then slowly poured into ice. Water was added, followed by ethyl acetate. After extraction, the organic layer was washed with aqueous sodium hydroxide (1%) and water, dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield 1-cyclopropyl-4-nitronaphthalene (5.2 g, 64%).

A solution of 1-cyclopropyl-4-nitronaphthalene (5 g, 23 mmol) in ethanol (200 mL) was stirred under hydrogen in the presence of Pd/C (10% net, 1.8 g). The reaction mixture was shaken overnight, filtered over celite, and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield 1-amino-4-cyclopropylnaphthalene (3.1 g, 73%).

Thiophosgene (1.1 g, 9.7 mmol) was added to a stirred solution of 1-amino-4-cyclopropylnaphthalene (1.8 g, 9.7 mmol) and diisopropylethylamine (2 eq) in dichloromethane (50 mL) at 0° C. The reaction mixture was stirred for 5 min at 0° C. and then aqueous HCl (1% solution) was added. The organic layer was separated, washed with brine, dried over sodium sulfate, filtered and the solvent removed under reduced pressure. Hexane was added, and the resulting precipitate was filtered. The solvent was evaporated to yield 1-cyclopropyl-4-isothiocyanatonaphthalene (1.88 g, 86%).
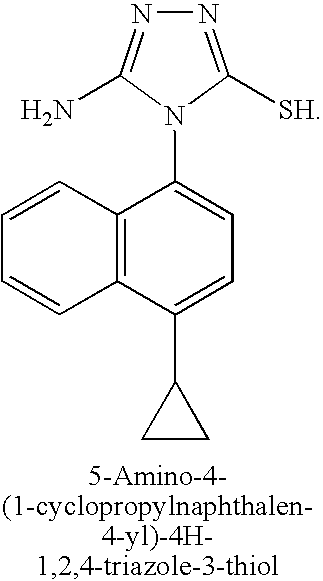
A mixture of aminoguanidine hydrochloride (3.18 g, 29 mmol), 1-cyclopropyl-4-isothiocyanatonaphthalene (3.24 g, 14 mmol) and diisopropylethylamine (3 eq) in DMF (20 mL) was stirred at 50° C. for 15 hours. The solvent was removed under reduced pressure, toluene added, and the solvent was evaporated again. Sodium hydroxide solution (2M, 30 mL) was added and the reaction mixture heated at 50° C. for 60 hours. The reaction mixture was filtered and the filtrate neutralized with aqueous HCl (2M). The mixture was re-filtered and the solvent removed under reduced pressure. The residue was purified by silica gel chromatography to yield 5-amino-4-(1-cyclopropylnaphthalen-4-yl)-4H-1,2,4-triazole-3-thiol (2.0 g, 49%).

Methyl 2-chloroacetate (0.73 mL, 8.3 mmol) was added dropwise over 5 mins to a suspension of 5-amino-4-(1-cyclopropylnaphthalen-4-yl)-4H-1,2,4-triazole-3-thiol (2.24 g, 7.9 mmol) and potassium carbonate (1.21 g, 8.7 mmol) in DMF (40 mL) at room temperature. The reaction was stirred at room temperature for 24 h and slowly poured into a stirred ice-cold water solution. The tan precipitate was collected by vacuum filtration and dried under high vacuum at 50° C. for 16 h in the presence of P2O5 to yield methyl 2-(5-amino-4-(1-cyclopropylnaphthalen-4-yl)-4H-1,2,4-triazol-3-ylthio)acetate (2.24 g, 80%).

Sodium nitrite (2.76 g, 40 mmol) was added to a solution of methyl 2-(5-amino-4-(1-cyclopropylnaphthalen-4-yl)-4H-1,2,4-triazol-3-ylthio)acetate (0.71 g, 2 mmol) and benzyltriethylammonium chloride (1.63 g, 6 mmol) in bromoform (10 mL). Dichloroacetic acid (0.33 mL, 4 mmol) was then added and the reaction mixture stirred at room temperature for 3 h. The mixture was directly loaded onto a 7-inch column of silica gel, packed with dichloromethane (DCM). The column was first eluted with DCM until all bromoform eluted, then eluted with acetone/DCM (5:95) to give methyl 2-(5-bromo-4-(1-cyclopropylnaphthalen-4-yl)-4H-1,2,4-triazol-3-ylthio)acetate (713 mg, 85%).
Example 104 Sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)acetate

Aqueous sodium hydroxide solution (1M, 2.0 mL, 2.0 mmol) was added dropwise over 5 mins to a solution of 2-(5-bromo-4-(1-cyclopropylnaphthalen-4-yl)-4H-1,2,4-triazol-3-ylthio)acetic acid (810 mg, 2.0 mmol) in ethanol (10 mL) at 10° C. The mixture was stirred at 10° C. for a further 10 mins. Volatile solvents were removed in vacuo to dryness to provide sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)acetate as a solid (850 mg, 100%).
Example 103 2-(5-Bromo-4-(1-cyclopropylnapthalen-4-yl)-4H-1,2,4-triazol-3-ylthio)acetic acid

A solution of lithium hydroxide (98 mg, 4.1 mmol) in water (10 mL) was added dropwise over 5 mins to a solution of methyl 2-(5-bromo-4-(1-cyclopropylnaphthalen-4-yl)-4H-1,2,4-triazol-3-ylthio)acetate (prepared as described in example 1 above; 1.14 g, 2.7 mmol) in ethanol (10 mL) and THF (10 mL) at 0° C. The mixture was stirred at 0° C. for a further 45 mins and then neutralized to pH 7 by the addition of 0.5N HCl solution at 0° C. The resulting mixture was concentrated in vacuo to ⅕th of its original volume, then diluted with water (˜20 mL) and acidified to pH 2-3 by the addition of 0.5N HCl to produce a sticky solid. (If the product comes out as an oil during acidification, extraction with DCM is recommended.) The tan solid was collected by vacuum filtration and dried under high vacuum at 50° C. for 16 h in the presence of P2O5 to yield 2-(5-bromo-4-(1-cyclopropylnaphthalen-4-yl)-4H-1,2,4-triazol-3-ylthio)acetic acid (1.02 g, 93%).
 |
1H NMR (400 MHz, DMSO-d6) δ ppm 0.84-0.91 (m, 2 H) 1.12-1.19 (m, 2 H) 2.54-2.61 (m, 1 H) 3.99 (d, J = 1.45 Hz, 2 H) 7.16 (d, J = 7.88 Hz, 1 H) 7.44 (d, J = 7.46 Hz, 1 H) 7.59-7.70 (m, 2 H) 7.75 (td, J = 7.62, 1.14 Hz, 1 H) 8.59 (d, J = 8.50 Hz, 1 H) 12.94 (br. s., 1 H) | Mass found: 404.5 (M + 1) | B |
……
POLYMORPHS AND SYNTHESIS
Described herein are various polymorphic, crystalline and mesophase forms of sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H-l,2,4-triazol-3-ylthio)acetate which decreases uric acid levels, (see for example US patent publication 2009/0197825, US patent publication 2010/0056464 and US patent publication 2010/0056465). Details of clinical studies involving sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H-l,2,4- triazol-3-ylthio)acetate have been described in International patent application
PCT/US2010/052958.
Polymorph Form A
In one embodiment, sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H- l,2,4-triazol-3-ylthio)acetate polymorph Form A exhibits an x-ray powder diffraction pattern characterized by the diffraction pattern summarized in Table 1 A or Table IB. In some embodiments, provided herein is a polymorph of sodium 2-(5-bromo-4-(4- cyclopropylnaphthalen-l-yl)-4H-l,2,4-triazol-3-ylthio)acetate comprising at least 3 peaks of (±0.1°2Θ) of Table 1A or IB. In certain embodiments, provided herein is a polymorph of sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H-l,2,4-triazol-3-ylthio)acetate comprising at least 4 peaks of (±0.1°2Θ) of Table 1A or IB, at least 5 peaks of (±0.1°2Θ) of Table 1A or IB, at least 6 peaks of (±0.1°2Θ) of Table 1A or IB, at least 8 peaks of
(±0. Γ2Θ) of Table 1A or IB, at least 10 peaks of (±0. Γ2Θ) of Table 1A, at least 15 peaks of (±0. Γ2Θ) of Table 1A, at least 20 peaks of (±0. Γ2Θ) of Table 1A, at least 25 peaks of (±0.1 °2Θ) of Table 1A, or at least 30 peaks of (±0.1 °2Θ) of Table 1A.


Examples
I Preparation of compounds
Example 1: Preparation of sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H-l,2,4- triazol-3-ylthio)acetate
Sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen- 1 -yl)-4H- 1 ,2,4-triazol-3-ylthio)acetate was prepared according to previously described procedures (see US patent publication
2009/0197825) and as outlined below.

[00103] Aqueous sodium hydroxide solution (1M, 2.0 mL, 2.0 mmol) was added dropwise over 5 min to a solution of 2-(5-bromo-4-(l-cyclopropylnaphthalen-4-yl)-4H- l,2,4-triazol-3-ylthio)acetic acid (810 mg, 2.0 mmol) in ethanol (10 mL) at 10 °C. The mixture was stirred at 10 °C for a further 10 min. Volatile solvents were removed in vacuo to dryness to provide sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H-l,2,4- triazol-3-ylthio)acetate as a solid (850 mg, 100%).
Example 2: Preparation of 2-(5-Bromo-4-(4-cyclopropylnaphthalen- 1 -yl)-4H- 1 ,2,4-triazol- 3-ylthio)acetic acid
2-(5-Bromo-4-(4-cyclopropylnaphthalen- 1 -yl)-4H- 1 ,2,4-triazol-3-ylthio)acetic acid was prepared according to previously described procedures (see US patent publication
2009/0197825) and as outlined below.
[00104] Route i:

Sodium hydroxide solution (2M aqueous, 33.7 mL, 67 mmol, 2 eq) was added to a suspension of 2-(5-bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H-l,2,4-triazol-3-ylthio)-N- (2-chloro-4-sulfamoylphenyl)acetamide (prepared by previously published procedures, see US 2009/0197825; 20 g, 34 mmol) in ethanol (200 mL) and the mixture heated at reflux for 4 hours. Charcoal (10 g) was added, the mixture stirred at room temperature for 12 hours and the charcoal removed by filtration. The charcoal was washed several times with ethanol and the filtrate then concentrated. Water (200 mL) was added and then concentrated to approx. one third volume to remove all ethanol. Water (200 mL) and ethyl acetate (250 mL) were added, the mixture stirred vigorously for 15 min and the organic layer removed. The aqueous layer was cooled to 0 °C and acidified by treatment with HCl (IN) resulting in the formation of a cloudy oily precipitate. The mixture was extracted with ethyl acetate (3x) and the combined organic extracts dried over sodium sulfate and concentrated to give 2-(5- bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H-l,2,4-triazol-3-ylthio)acetic acid as an off white solid (11.2 g, 82%).
[00105] Route ii:

STEP A: 1-Cyclopropylnaphthalene

Cyclopropylmagnesium bromide (150 mL, 0.5M in tetrahydrofuran) was slowly added to a solution of 1-bromonaphthalene (10 g, 50 mmol) and [l,3-bis(diphenylphosphino)propane] dichloro nickel (II) in tetrahydrofuran (10 mL) stirred at 0 °C, and the reaction mixture stirred at room temperature for 16 hours. The solvent was removed under reduced pressure and ethyl acetate and aqueous ammonium chloride were added. After extraction, the organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield 1-cyclopropylnaphthalene (6.4 g, 76%). ] STEP B: l-Cyclopropyl-4-nitronaphthalene

Sodium nitrite (30 mL) was slowly added (over 2 hours) to 1-cyclopropylnaphthalene (6.4 g, 38 mmol) stirred at 0 °C. The reaction mixture was stirred at 0 °C for an extra 30 min and then slowly poured into ice. Water was added, followed by ethyl acetate. After extraction, the organic layer was washed with aqueous sodium hydroxide (1%) and water, dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield l-cyclopropyl-4-nitronaphthalene (5.2 g, 64%).
[00108] STEP C: l-Amino-4-cyclopropylnaphthalene

A solution of l-cyclopropyl-4-nitronaphthalene (5 g, 23 mmol) in ethanol (200 mL) was stirred under hydrogen in the presence of Pd/C (10% net, 1.8 g). The reaction mixture was shaken overnight, filtered over celite, and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield l-amino-4-cyclopropylnaphthalene (3.1 g, 73%).
STEP D: l-Cyclopropyl-4-isothiocvanatonaphthalene

Thiophosgene (1.1 g, 9.7 mmol) was added to a stirred solution of l-amino-4- cyclopropylnaphthalene (1.8 g, 9.7 mmol) and diisopropylethylamine (2 eq) in
dichloromethane (50 mL) at 0 °C. The reaction mixture was stirred for 5 min at 0 °C and then aqueous HCl (1% solution) was added. The organic layer was separated, washed with brine, dried over sodium sulfate, filtered and the solvent removed under reduced pressure. Hexane was added, and the resulting precipitate was filtered. The solvent was evaporated to yield l-cyclopropyl-4-isothiocyanatonaphthalene (1.88 g, 86%>).
[00110] STEP E: 5-Amino-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4-triazole-3- thiol

A mixture of aminoguanidine hydrochloride (3.18 g, 29 mmol), l-cyclopropyl-4- isothiocyanato naphthalene (3.24 g, 14 mmol) and diisopropylethylamine (3 eq) in DMF (20 mL) was stirred at 50 °C for 15 hours. The solvent was removed under reduced pressure, toluene added, and the solvent was evaporated again. Sodium hydroxide solution (2M, 30 mL) was added and the reaction mixture heated at 50 °C for 60 hours. The reaction mixture was filtered and the filtrate neutralized with aqueous HCl (2M). The mixture was re-filtered and the solvent removed under reduced pressure. The residue was purified by silica gel chromatography to yield 5-amino-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4-triazole-3- thiol (2.0 g, 49%). [00111] STEP F: Methyl 2-(5-amino-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4- -3 -ylthio)acetate

Methyl 2-chloroacetate (0.73 mL, 8.3 mmol) was added dropwise over 5 min to a suspension of 5-amino-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4-triazole-3-thiol (2.24 g, 7.9 mmol) and potassium carbonate (1.21 g, 8.7 mmol) in DMF (40 mL) at room
temperature. The reaction was stirred at room temperature for 24 h and slowly poured into a stirred ice-cold water solution. The tan precipitate was collected by vacuum filtration and dried under high vacuum at 50 °C for 16 h in the presence of P2O5 to yield methyl 2-(5- amino-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4-triazol-3-ylthio)acetate (2.24 g, 80%).
[00112] STEP G: Methyl 2-(5-bromo-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4- triazol-3 -ylthio)acetate

Sodium nitrite (2.76 g, 40 mmol) was added to a solution of methyl 2-(5-amino-4-(l- cyclopropylnaphthalen-4-yl)-4H-l,2,4-triazol-3-ylthio)acetate (0.71 g, 2 mmol) and benzyltriethylammonium chloride (1.63 g, 6 mmol) in bromoform (10 mL). Dichloroacetic acid (0.33 mL, 4 mmol) was then added and the reaction mixture stirred at room
temperature for 3 h. The mixture was directly loaded onto a 7-inch column of silica gel, packed with dichloromethane (DCM). The column was first eluted with DCM until all bromoform eluted, then eluted with acetone/DCM (5:95) to give methyl 2-(5-bromo-4-(l- cyclopropylnaphthalen-4-yl)-4H-l,2,4-triazol-3-ylthio)acetate (713 mg, 85%).
[00113] STEP H: 2-(5-Bromo-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4-triazol-3- )acetic acid

A solution of lithium hydroxide (98 mg, 4.1 mmol) in water (10 mL) was added dropwise over 5 min to a solution of methyl 2-(5-bromo-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4- triazol-3-ylthio)acetate (1.14 g, 2.7 mmol) in ethanol (10 mL) and THF (10 mL) at 0 °C. The mixture was stirred at 0 °C for a further 45 min and then neutralized to pH 7 by the addition of 0.5N HC1 solution at 0 °C. The resulting mixture was concentrated in vacuo to l/5th of its original volume, then diluted with water (~20 mL) and acidified to pH 2-3 by the addition of 0.5N HC1 to produce a sticky solid. (If the product comes out as an oil during acidification, extraction with dichloromethane is recommended.) The tan solid was
collected by vacuum filtration and dried under high vacuum at 50 °C for 16 h in the
presence of P2O5 to yield 2-(5-bromo-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4-triazol-3- ylthio)acetic acid (1.02 g, 93%).
………………………….
EXAMPLES
The following experiments are provided only by way of example, and should not be understood as limiting the scope of the invention.
COMPOUNDS OF THE INVENTION 2-[5-Bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]-N-(2-chloro-4-sulfamoylphenyl)acetamide (Method A)

1-Cyclopropyl-naphthalene
Cyclopropylmagnesium bromide (150 mL, 0.5 M in tetrahydrofuran) was slowly added to a solution of 1-bromo-naphthalene (10 g, 50 mmol) and [1,3-bis(diphenylphosphino)propane]dichloronickel(II) in tetrahydrofuran (10 mL) stirred at 0° C. The reaction mixture was stirred at room temperature for 16 hours and the solvent was evaporated under reduced pressure. EtOAc and ammonium chloride in water were added. After extraction, the organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield 1-cyclopropyl-naphthalene (6.4 g, 76%).
1-Cyclopropyl-4-nitro-naphthalene
Sodium nitrite (30 mL) was slowly added (over 2 hours) to 1-cyclopropyl-naphthalene (6.4 g, 38 mmol) stirred at 0° C. The reaction mixture was stirred at 0° C. for an extra 30 min and then was slowly poured into ice. Water was added, followed by EtOAc. After extraction, the organic layer was washed with a 1% aqueous solution of NaOH, then washed with water, dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield 1-cyclopropyl-4-nitro-naphthalene (5.2 g, 64%).
1-Amino-4-cyclopropyl-naphthalene
A solution of 1-cyclopropyl-4-nitro-naphthalene (5 g, 23 mmol) in ethanol (200 mL) was stirred under hydrogen in the presence of Pd/C (10% net, 1.8 g). The reaction mixture was shaken overnight, then filtered over celite. The solvent was evaporated, and the residue was purified by silica gel chromatography to yield 1-amino-4-cyclopropyl-naphthalene (3.1 g, 73%).
1-Cyclopropyl-4-isothiocyanato-naphthalene
Thiophosgene (1.1 g, 9.7 mmol) was added to a solution of 1-amino-4-cyclopropyl-naphthalene (1.8 g, 9.7 mmol) and diisopropylethylamine (2 eq) in dichloromethane (50 mL) stirred at 0° C. The reaction mixture was stirred for 5 min at this temperature, then a 1% solution of HCl in water was added and the organic layer was separated, washed with brine, dried over sodium sulfate, filtered and the solvent was evaporated under reduced pressure. Hexane was added, and the resulting precipitate was filtered. The solvent was evaporated to yield 1-cyclopropyl-4-isothiocyanatonaphthalene (1.88 g, 86%).
5-Amino-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazole-3-thiol
A mixture of aminoguanidine hydrochloride (3.18 g, 29 mmol), 1-cyclopropyl-4-isothiocyanato-naphthalene (3.24 g, 14 mmol) and diisopropylethylamine (3 eq) in DMF (20 mL) was stirred at 50° C. for 15 hours. The solvent was evaporated, toluene was added, and the solvent was evaporated again. A 2.0 M aqueous solution of sodium hydroxide (30 mL) was added and the reaction mixture was heated at 50° C. for 60 hours. The reaction mixture was filtered, and the filtrate was neutralized with a 2.0 M aqueous solution of HCl. New filtration, then evaporation of solvent and purification of the residue by silica gel chromatography to yield 5-amino-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazole-3-thiol (2.0 g, 49%).
2-[5-Amino-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]-N-(2-chloro-4-sulfamoylphenyl)Acetamide
In a solution of 5-amino-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazole-3-thiol (708 mg, 2.5 mmol), K2CO3 (380 mg, 2.5 mmol) in DMF (20 mL) was added 2-chloro-N-(2-chloro-4-sulfamoylphenyl)acetamide (710 mg, 2.5 mmol). The reaction mixture was stirred at room temperature overnight. Upon completion of the reaction, the solvent was evaporated. The residue was purified by silica gel chromatography to yield 2-[5-Amino-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]-N-(2-chloro-4-sulfamoylphenyl)acetamide (1.26 g, 95%).
2-[5-Bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]-N-(2-chloro-4-sulfamoylphenyl)acetamide
Dichloroacetic acid (180 uL, 2.2 mmol) was added to a suspension of 2-[5-amino-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]-N-(2-chloro-4-sulfamoylphenyl)acetamide (0.59 g, 1.1 mmol), sodium nitrite (1.5 g, 22 mmol) and BTEABr (0.91 g, 3.3 mmol) in dibromomethane (30 mL). The reaction mixture was stirred at room temperature for 4 hours, then extracted with dichloromethane and sodium bicarbonate in water. The organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield 2-[5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]-N-(2-chloro-4-sulfamoylphenyl)acetamide (224 mg, 31%).
2-[5-Bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazole-3-ylsulfanyl]-N-(2-chloro-4-sulfamoylphenyl)acetamide (Method B)

2-[5-Amino-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]acetic acid methyl ester
| Materials | Amount | Mol. Wt. | mmoles | |
| thiotriazole | 2.24 | g | 282.36 | 7.9 |
| methyl chloroacetate | 0.73 | ml | 108.52 | 8.3 (1.05 eq) |
| potassium carbonate | 1.21 | g | 138.21 | 8.7 (1.1 eq) |
| dimethylformamide | 40 | ml | (5 mL/mmol) | |
Procedure:
To a suspension of thiotriazole and potassium carbonate in DMF was added methyl chloroacetate dropwise at room temperature for 5 min. The reaction was stirred at room temperature for 24 h and slowly poured into a stirred ice-cold water solution. The tan precipitate was collected by vacuum filtration and dried under high vacuum at 50° C. for 16 h in the presence of P2O5 to yield 2.24 g (80%) of the title compound.
2-[5-Bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]acetic acid methyl ester
| Materials | Amount | Mol. Wt. | mmoles | ||
| thiotriazole L10183-58 | 709 | mg | 354.43 | 2.0 | |
| bromoform | 10 | ml | (5 ml/mmol) | ||
| sodium nitrite | 2.76 | g | 69.00 | 40 | (20 eq) |
| benzyltriethylammonium | 1.63 | g | 272.24 | 6.0 | (3 eq) |
| bromide | |||||
| dichloroacetic acid | 0.33 | ml | 128.94 | 4.0 | (2 eq) |
Procedure:
To a solution of 2-[5-amino-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]acetic acid methyl ester and benzyltriethylammonium chloride in bromoform was added sodium nitrite. To the mixture was added dichloroacetic acid and the reaction mixture was stirred at room temperature for 3 h. The mixture was directly loaded onto a 7-inch column of silica gel that was packed with CH2Cl2. The column was first eluted with CH2Cl2 until all CHBr3 eluted, and was then eluted with acetone/CH2Cl2 (5:95) to give 713 mg (85%) of the title compound.
2-[5-Bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]acetic acid
| Materials | Amount | Mol. Wt. | mmoles | |
| thiotriazole methyl ester | 1.14 | g | 418.31 | 2.7 |
| tetrahydrofuran | 10 | ml | (~3 ml/mmol) | |
| ethanol | 10 | ml | (~3 ml/mmol) | |
| water | 10 | ml | (~3 ml/mmol) | |
| lithium hydroxide | 98 | mg | 23.95 | 4.1 (1.5 eq) |
Procedure:
To a solution of 2-[5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]acetic acid methyl ester, in a mixture of THF and EtOH at 0° C., was added a solution of LiOH in H2O dropwise over 5 min. The reaction was complete after stirring at 0° C. for an additional 45 min. The reaction was neutralized to pH 7 by the addition of 0.5 N HCl solution at 0° C., and the resulting mixture was concentrated in vacuo to ⅕th of its original volume. The mixture was diluted with H2O (˜20 mL) and acidified to pH 2-3 by the addition of 0.5 N HCl to produce sticky solid. (If the product comes out as an oil during acidification, extraction with CH2Cl2 is recommended.) The tan solid was collected by vacuum filtration and dried under high vacuum at 50° C. for 16 h in the presence of P2O5 to yield 1.02 g (93%) of the title compound.
REF:
Esmir Gunic, Jean-Luc Girardet, Jean-Michel Vernier, Martina E. Tedder, David A. Paisner;Compounds, compositions and methods of using same for modulating uric acid levels;US patent number US8242154 B2 ;Also published as US20100056465, US20130040907;Original Assignee: Ardea Biosciences, Inc
Esmir Gunic, Jean-Luc Girardet, Jean-Michel Vernier, Martina E. Tedder, David A. Paisner;Compounds, compositions and methods of using same for modulating uric acid levels;US patent number US8173690 B2;Also published as US20100056464;Original Assignee: Ardea Biosciences, Inc
Barry D. Quart, Jean-Luc Girardet, Esmir Gunic, Li-Tain Yeh;Compounds and compositions and methods of use;US patent number US8084483 B2; Also published as CA2706858A1, CA2706858C, CN101918377A, CN102643241A, CN103058944A, EP2217577A2, EP2217577A4, US8283369, US8357713, US8546437, US20090197825, US20110268801, US20110293719, US20120164222, US20140005136, WO2009070740A2, WO2009070740A3;Original Assignee:Ardea Biosciences, Inc.
Gunic, Esmir; Galvin, Gabriel;Manufacture of 2-[5-bromo-4-(cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio]acetic acid and related compounds;PCT Int. Appl., WO2014008295 A1
Zamansky, Irina et al;Process for preparation of polymorphic, crystalline, and mesophase forms of 2-[[5-bromo-4-(4-cyclopropyl-1-naphthalenyl)-4H-1,2,4-triazol-3-yl]thio]acetic acid sodium salt; PCT Int. Appl., WO2011085009
Gunic, Esmir et al;Preparation of naphthalene thio triazole derivatives and their use for modulating uric acid levels; U.S. Pat. Appl. Publ., 20100056465
unic, Esmir et al;Preparation of naphthalene thio triazole derivatives and their use for modulating uric acid levels;U.S. Pat. Appl. Publ., 20100056464
Quart, Barry D. et al;Preparation of azole carboxylates as modulators of blood uric acid levels;PCT Int. Appl., 2009070740, 04 Jun 2009
Girardet, Jean-Luc et al;Preparation of S-triazolyl α-mercaptoacetanilides as inhibitors of HIV reverse transcriptase;PCT Int. Appl., WO2006026356
US20100056465 * Sep 4, 2009 Mar 4, 2010 Ardea Biosciences Compounds, compositions and methods of using same for modulating uric acid levels
US20100056542 * Sep 4, 2009 Mar 4, 2010 Ardea Biosciences Compounds, compositions and methods of using same for modulating uric acid levels
WO2009070740A2 * Nov 26, 2008 Jun 4, 2009 Ardea Biosciences Inc Novel compounds and compositions and methods of use
WO2011085009A2 * Jan 5, 2011 Jul 14, 2011 Ardea Biosciences, Inc. Polymorphic, crystalline and mesophase forms of sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4h-1,2,4-triazol-3-ylthio)acetate, and uses thereof
| WO2011159732A1 * | Jun 14, 2011 | Dec 22, 2011 | Ardea Biosciences,Inc. | Treatment of gout and hyperuricemia |
| WO2012092395A2 * | Dec 28, 2011 | Jul 5, 2012 | Ardea Biosciences, Inc. | Polymorphic forms of 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4h-1,2,4-triazol-3-ylthio) acetic acid and uses thereof |
| EP2560642A2 * | Mar 29, 2011 | Feb 27, 2013 | Ardea Biosciences, Inc. | Treatment of gout |
| US8546436 | Dec 28, 2011 | Oct 1, 2013 | Ardea Biosciences, Inc. | Polymorphic forms of 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)acetic acid and uses thereof |
| US20100056465 * | Sep 4, 2009 | Mar 4, 2010 | Ardea Biosciences | Compounds, compositions and methods of using same for modulating uric acid levels |
| US20100056542 * | Sep 4, 2009 | Mar 4, 2010 | Ardea Biosciences | Compounds, compositions and methods of using same for modulating uric acid levels |
| WO2009070740A2 * | Nov 26, 2008 | Jun 4, 2009 | Ardea Biosciences Inc | Novel compounds and compositions and methods of use |
| WO2011085009A2 * | Jan 5, 2011 | Jul 14, 2011 | Ardea Biosciences, Inc. | Polymorphic, crystalline and mesophase forms of sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4h-1,2,4-triazol-3-ylthio)acetate, and uses thereof |
| US8283369 | 30 Jun 2011 | 9 Oct 2012 | Ardea Biosciences. Inc. | Compounds and compositions and methods of use |
| US8357713 | 30 Jun 2011 | 22 Jan 2013 | Ardea Biosciences Inc. | Compounds and compositions and methods of use |
| US8546437 | 30 Jun 2011 | 1 Oct 2013 | Ardea Biosciences, Inc. | Compounds and compositions and methods of use |
| US8633232 * | 4 May 2012 | 21 Jan 2014 | Ardea Biosciences, Inc. | Compounds, compositions and methods of using same for modulating uric acid levels |
| US20130040907 * | 4 May 2012 | 14 Feb 2013 | Ardea Biosciences Inc. | Compounds, Compositions and Methods of Using Same for Modulating Uric Acid Levels |
| WO2006026356A2 * | Aug 25, 2005 | Mar 9, 2006 | La Rosa Martha De | S-TRIAZOLYL α-MERCAPTOACETANILDES AS INHIBITORS OF HIV REVERSE TRANSCRIPTASE |
| WO2009070740A2 * | Nov 26, 2008 | Jun 4, 2009 | Ardea Biosciences Inc | Novel compounds and compositions and methods of use |
| US20090197825 * | Nov 26, 2008 | Aug 6, 2009 | Ardea Biosciences, Inc. | Novel compounds and compositions and methods of use |
| US7947721 | Aug 25, 2005 | May 24, 2011 | Ardes Biosciences Inc. | S-triazolyl α-mercaptoacetanilides as inhibitors of HIV reverse transcriptase |
| US8084483 | Nov 26, 2008 | Dec 27, 2011 | Ardea Biosciences, Inc. | Compounds and compositions and methods of use |
| US8106205 | Feb 2, 2010 | Jan 31, 2012 | Ardea Biosciences, Inc. | N[S(4-aryl-triazol-3-yl)α-mercaptoacetyl]p-amino benzoic acids as HIV reverse transcriptase inhibitors |
| US8252828 | Jun 30, 2011 | Aug 28, 2012 | Ardea Biosciences, Inc. | S-triazolyl α-mercapto acetanilides as inhibitors of HIV reverse transcriptase |
| US8283369 | Jun 30, 2011 | Oct 9, 2012 | Ardea Biosciences. Inc. | Compounds and compositions and methods of use |
| US8357713 | Jun 30, 2011 | Jan 22, 2013 | Ardea Biosciences Inc. | Compounds and compositions and methods of use |
| US8372807 | May 20, 2010 | Feb 12, 2013 | Ardea Biosciences, Inc. | Methods of modulating uric acid levels |
| US8481581 | Jul 18, 2012 | Jul 9, 2013 | Ardea Biosciences, Inc. | S-triazolyl α-mercaptoacetanilides as inhibitors of HIV reverse transcriptase |
| US8524754 | Jan 5, 2011 | Sep 3, 2013 | Ardea Biosciences, Inc. | Polymorphic, crystalline and mesophase forms of sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio) acetate, and uses thereof |
| US8546436 | Dec 28, 2011 | Oct 1, 2013 | Ardea Biosciences, Inc. | Polymorphic forms of 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)acetic acid and uses thereof |
| US8546437 | Jun 30, 2011 | Oct 1, 2013 | Ardea Biosciences, Inc. | Compounds and compositions and methods of use |

An unexpected phenomenon concerning an otherwise common impurity of lesinurad has been observed in the context of synthetic process development. A new industrial process was designed as a chlorine-free process, but the critical chlorinated impurity 10 was surprisingly detected in the isolated product. Because of the structural similarity of the impurity and the product, no efficient separation of 10 by conventional methods (e.g., crystallization) was discovered. The formation of the impurity was explained by a chlorine impurity in a commercial brominating agent. This communication also describes control of the critical impurity.
Identification of an Unexpected Impurity in a New Improved Synthesis of Lesinurad

2-[[5-Bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-yl]thio]acetic Acid (1)
1H NMR (DMSO-d6), δ (ppm): 0.87 (m, 2H); 1.15 (m, 2H); 2.56 (m, 1H); 4.00 (m, 2H); 7.16 (d, J = 8.5 Hz, 1H); 7.44 (d, J = 7.3, 1H); 7.64 (d, J = 7.3 Hz, 1H); 7.66 (t, J= 7.6 Hz, 1H); 7.74 (t, J = 7.6 Hz, 1H); 8.58 (d, J = 8.5 Hz, 1H); 12.99 (s, COOH). 13C NMR (DMSO-d6), δ (ppm): 7.3; 7.4; 12.9; 34.1; 121.8; 122.7; 125.2; 126.6; 126.8; 127.3; 128.1; 128.6; 131.4; 133.5; 143.2; 153.5; 169.0.
