
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
FDA approves first retinal implant for adults with rare genetic eye disease
 Argus II (credit: Second Sight)
Argus II (credit: Second Sight)Second Sight Medical Products, Inc., is the manufacturer and is already approved in Europe

Argus II components (credit: FDA)
The Argus II design consists of an external video camera system matched to the implanted retinal stimulator, which contains a microelectrode array that spans 20 degrees of visual field.
An external camera system, built into a pair of glasses, streams video to a belt-worn computer, which converts the video into stimulus commands for the implant.
The belt-worn video processing unit (computer) encodes the commands into a wireless signal that is transmitted to the implant, which has the necessary electronics to receive and decode both wireless power and data.
Artificial retina device, consisting of a glasses-mounted camera and a microchip surgically implanted on the retina
Based on those data, the implant stimulates the retina with small electrical pulses. The electronics are hermetically packaged and the electrical stimulus is delivered to the retina via a microelectrode array.
Peregrine Pharmaceuticals Announces Results From Phase II Clinical Trial of Bavituximab in Stage IV Pancreatic Cancer

TUSTIN, CA 02/13/13 — Peregrine Pharmaceuticals announced results from its 70 patient open-label, randomized Phase II clinical trial of bavituximab used in combination with gemcitabine in patients with previously untreated, advanced Stage IV pancreatic cancer. The trial included the enrollment of patients with advanced metastatic disease including significant liver involvement and poor performance status associated with rapid disease progression. Results showed that the combination of bavituximab and gemcitabine resulted in more than a doubling of overall response rates (ORR) and an improvement in overall survival (OS) when compared with gemcitabine alone (control arm). In the trial, patients treated with a combination of bavituximab and gemcitabine had a 28% tumor response rate as compared to 13% in the control arm. Median OS, the primary endpoint of the trial, was 5.6 months for the bavituximab plus gemcitabine arm and 5.2 months for the control arm (hazard ratio = 0.75).
Bavituximab binds to phosphatidylserine which is exposed on the surface of certain atypical animal cells, including tumour cells and cells infected with any of six different families of virus. These viral families contain the viruses hepatitis C, influenza A and B, HIV 1 and 2, measles, respiratory syncytial virus and pichinde virus, which is a model for the deadly Lassa virus.[2] Other cells are not affected since phosphatidylserine normally is only intracellular.[3]

Bavituximab binds to various aminophospholipids and is dependent on interaction with plasma protein beta-2 glycoprotein I to mediate binding.
These target aminophospholipids, usually residing only on the inner leaflet of the plasma membrane of cells, become exposed in virally infected, damaged or malignant cells, and more generally in most cells undergoing the process of apoptosis.
The antibody’s binding to phospholipids alerts the body’s immune system to attack the tumor endothelial cells, thrombosing the tumor’s vascular network and/or attacking free floating virally infected and metastatic cells while potentially minimizing side effects in healthy tissues.
- Statement on a nonproprietary name adopted by the USAN council
- Nature Medicine 14, 1357 – 1362 (2008)
- He, J.; Yin, Y.; Luster, T. A.; Watkins, L.; Thorpe, P. E. (2009). “Antiphosphatidylserine Antibody Combined with Irradiation Damages Tumor Blood Vessels and Induces Tumor Immunity in a Rat Model of Glioblastoma”. Clinical Cancer Research 15 (22): 6871–6880. doi:10.1158/1078-0432.CCR-09-1499. PMID 19887482. edit
- New Progression-Free Survival Data From Peregrine’s Bavituximab in Phase II Refractory Breast Cancer
- Phase II Advanced Breast Cancer Data to Be Presented at ASCO Highlight Promising Tumor Response and Progression-Free Survival Data With Peregrine’s Bavituximab
- Pharma company completes humanization of 3G4 antibody
- He, J.; Luster, T. A.; Thorpe, P. E. (2007). “Radiation-Enhanced Vascular Targeting of Human Lung Cancers in Mice with a Monoclonal Antibody That Binds Anionic Phospholipids”. Clinical Cancer Research 13 (17): 5211–5218. doi:10.1158/1078-0432.CCR-07-0793. PMID 17785577. edit
- Ran; Downes, A.; Thorpe, P. E. (2002). “Increased exposure of anionic phospholipids on the surface of tumor blood vessels”. Cancer Research 62 (21): 6132–6140. PMID 12414638.
<a href=”http://www.bloglovin.com/blog/4758019/?claim=ukqfxgh6tk3″>Follow my blog with Bloglovin</a>
AbbVie Announces First Long-term, Patient-Reported Health Outcomes Data for Use of HUMIRA® (Adalimumab) in Patients with Pediatric Crohn’s Disease
Adalimumab
monoclonal antibody
http://www.nature.com/nrd/journal/v2/n9/fig_tab/nrd1182_F1.html
VIENNA, Feb. 15, 2013 AbbVie announced the first long-term, patient-reported health outcomes data from analyses of the Phase 3 IMAgINE-1 trial. The analyses assessed improvements in health-related quality of life (HRQOL) measures for pediatric patients aged 6 to 17 years with severe active Crohn’s disease, taking HUMIRA, who had an inadequate response, were intolerant or had contraindications to conventional therapy, as well as the work productivity of their caregivers throughout the 52-week study. The results of these analyses are being presented this week at the European Crohn’s and Colitis Organisation (ECCO) 8th Annual Congress.
Adalimumab (HUMIRA, Abbott) is the third TNF inhibitor, after infliximab and etanercept, to be approved in the United States. Like infliximab and etanercept, adalimumab binds to Tumor necrosis factor-alpha (TNFα), preventing it from activating TNF receptors. Adalimumab was constructed from a fully human monoclonal antibody, while infliximab is a mouse-human chimeric antibody and etanercept is a TNF receptor-IgG fusion protein. TNFα inactivation has proven to be important in downregulating the inflammatory reactions associated with autoimmune diseases. As of 2008 adalimumab has been approved by the U.S. Food and Drug Administration (FDA) for the treatment of rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis, Crohn’s disease, moderate to severe chronic psoriasis and juvenile idiopathic arthritis. Although only approved for ulcerative colitis from late 2012 by the FDA in the disease’s management, it has been used for several years in cases that have not responded to conventional treatment at standard dosing for Crohn’s Disease.

However, because TNFα is part of the immune system that protects the body from infection, prolonged treatment with adalimumab may slightly increase the risk of developing infections.
HUMIRA (“Human Monoclonal Antibody in Rheumatoid Arthritis”) is marketed in both preloaded 0.8 mL syringes and also in preloaded pen devices (called Humira Pen), both injected subcutaneously, typically by the patient at home.
Adalimumab was discovered as a result of the collaboration between BASF Bioresearch Corporation (Worcester, Massachusetts, a unit of BASF) and Cambridge Antibody Technology which began in 1993.[4]
The drug candidate was discovered initially using CAT’s phage display technology and named D2E7.[2] The key components of the drug were found by guiding the selection of human antibodies from phage display repertoires to a single epitope of an antigen TNF alpha.[5] The ultimate clinical candidate, D2E7, was created and manufactured at BASF Bioresearch Corporation and taken through most of the drug development process by BASF Knoll, then further development, manufacturing and marketing by Abbott Laboratories, after Abbott acquired the pharmaceutical arm of BASF Knoll.[6]
Adalimumab was the first fully human monoclonal antibody drug approved by the FDA. It was derived from phage display,[1] and was discovered through a collaboration between BASF Bioresearch Corporation (Worcester, Massachusetts, a unit of BASF) and Cambridge Antibody Technology as D2E7,[2] then further manufactured at BASF Bioresearch Corporation and developed by BASF Knoll (BASF Pharma) and, ultimately, manufactured and marketed by Abbott Laboratories after the acquisition of BASF Pharma by Abbott.
In 2009, HUMIRA had over $5 billion in annual sales.[3]

Components of a Humira autoinjector pen
- Brekke OH , Sandlie I (January 2003). “Therapeutic antibodies for human diseases at the dawn of the twenty-first century”. Nat Rev Drug Discov 2 (1): 52–62. doi:10.1038/nrd984. PMID 12509759.
- Kempeni J (January 1999). “Preliminary results of early clinical trials with the fully human anti-TNFα monoclonal antibody D2E7”. Ann Rheum Dis 58 (suppl 1): I70–2. doi:10.1136/ard.58.2008.i70. PMC 1766582. PMID 10577977.
- http://www.abbott.com/static/content/microsite/annual_report/2006/humira.html
- [1] Cambridge Antibody Technology website
- Jespers LS, Roberts A, Mahler SM, Winter G, Hoogenboom HR (September 1994). “Guiding the selection of human antibodies from phage display repertoires to a single epitope of an antigen”. Biotechnology (N.Y.) 12 (9): 899–903. doi:10.1038/nbt0994-899. PMID 7521646.
- http://www2.basf.us/corporate/news2000/newsknoll_pharma_121500.html
- http://www.prnewswire.com/cgi-bin/stories.pl?ACCT=104&STORY=/www/story/11-12-2001/0001613559&EDATE=
- Rau R (January 2002). “Adalimumab (a fully human anti-tumour necrosis factor α monoclonal antibody) in the treatment of active rheumatoid arthritis: the initial results of five trials”. Ann Rheum Dis 61 (Suppl 2): ii70–3. doi:10.1136/ard.61.suppl_2.ii70. PMC 1766697. PMID 12379628.

Genzyme Announces Positive New Data from Two Phase 3 Studies for Oral Eliglustat Tartrate for Gaucher Disease
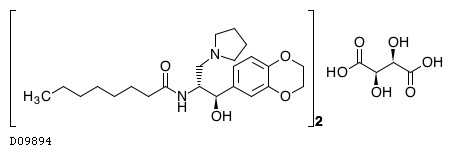
Eliglustat tartrate (USAN)
ENGAGE Study Results:
In ENGAGE, a Phase 3 trial to evaluate the safety and efficacy of eliglustat tartrate in 40 treatment-naïve patients with Gaucher disease type 1, improvements were observed across all primary and secondary efficacy endpoints over the 9-month study period. Results were reported today at the WORLD Symposium by Pramod Mistry, MD, PhD, FRCP, Professor of Pediatrics & Internal Medicine at Yale University School of Medicine, and an investigator in the trial.
The randomized, double-blind, placebo-controlled study had a primary efficacy endpoint of improvement in spleen size in patients treated with eliglustat tartrate. Patients were stratified at baseline by spleen volume. In the study, a statistically significant improvement in spleen size was observed at nine months in patients treated with eliglustat tartrate compared with placebo. Spleen volume in patients treated with eliglustat tartrate decreased from baseline by a mean of 28 percent compared with a mean increase of two percent in placebo patients, for an absolute difference of 30 percent (p<0.0001).
Eliglustat tartate (Genz-112638)
What is Eliglustat?
- Eliglustat is a new investigational phase 3 compound from Genzyme Corporation that is being studied for type 1 Gaucher Disease.
- Eliglustat works as a substrate reduction therapy by reducing glucocerebroside. formation.
- This product is an oral agent (i.e. a pill) that is taken once or twice a day in contrast to an IV infusion for enzyme replacement therapy. Enzyme replacement therapy focuses on replenishing the enzyme that is deficient in Gaucher Disease and breaks down glucocerebroside that accumulates.
- The clinical trials for eliglustat tartate are sponsored by Genzyme Corporation.


BioAlliance Pharma announces the forthcoming extension of its phase II clinical trial with Validive® in the United States

CLONIDINE
FEB14 2013
BioAlliance Pharma SA (Euronext Paris – BIO), an innovative Company dedicated to the development of orphan oncology products and to supportive care products, announces the extension of its phase II clinical trial with Validive® (clonidine Lauriad™) in the United States in radio/chemotherapy-induced oral mucositis prevention in patients with head and neck cancer.
Further to approval by the United States FDA (Food and Drug Administration), BioAlliance Pharma will extend its clinical trial to the United States, increasing the number of clinical investigation centers involved in this randomized double blind phase II trial.
So far almost 50% of planned patients have been enrolled in about 30 European centers. With the upcoming initiation of several centers in the United States, BioAlliance Pharma expects to finalize trial recruitment in early 2014 with results expected the same year.
“Beyond accelerating recruitment, the extension of the trial to the United States is also a key factor to reinforce our international panel of scientific experts and clinical investigators around Validive®. This will raise awareness and create hands-on experience of the drug of future key prescribers of Validive® in major US centers specialized in oncology and radiotherapy,” stated Judith Greciet, CEO of BioAlliance Pharma.
Severe oral mucositis is a particularly invalidating pathology occurring in more than 60% of patients treated with radio/chemotherapy for head and neck cancer and has currently no validated curative or preventive treatment. It may induce intense oral pain and eating disability requiring enteral or parenteral nutritional support. Thirty per cent of patients need to be hospitalized as a result and symptoms can force patients to stop treatment for an undefined period thus reducing treatment efficacy.
Clonidine is a sympatholytic medication used to treat medical conditions, such as high blood pressure, ADHD, anxiety/panic disorder, and certain pain conditions. It is classified as a centrally acting α2 adrenergic agonist. An alternative hypothesis that has been proposed is that clonidine acts centrally as an imidazoline receptor agonist.
Synthesis
Clonidine, 2-(2,6-dichlorophenylamino)imidazoline, is synthesized from 2,6-dichloroaniline, the reaction of which with ammonium thiocyanate gives N-(2,6- dichlorophenyl)thiourea. Methylation of this product, followed by the subsequent reaction with ethylene diamine gives clonidine. 
- K. Zeile, H. Staehle, K. H. Hauotman, DE 1303141 (1961).
- H. Stahle, K. Zeile, U.S. Patent 3,202,660 (1965).
- K. Zeile, K. H. Hauotman, H. Stahle, U.S. Patent 3,236,857 (1966).
- Boehringer Sohn Ingelheim, BE 653933 (1964).
- Boehringer Sohn Ingelheim, GB 1016514 (1962).
- Boehringer Ingelheim GmbH, GB 1034938 (1964).

US patent 3937717, Stahle, H.; Koppe, H.; Kummer, W.; Stockhaus, K., “2-Phenylamino-imidazolines-(2)”, issued 1976-02-10, assigned to Boehringer Ingelheim GmbH
CLORPRES (clonidine hydrochloride and chlorthalidone) ® is a combination of clonidine hydrochloride (a centrally acting antihypertensive agent) and chlorthalidone (a diuretic).
CLORPRES (clonidine hydrochloride and chlorthalidone) ® is available as tablets for oral administration in three dosage strengths: 0.1 mg/15 mg, 0.2 mg/15 mg and 0.3 mg/15 mg of clonidine hydrochloride/chlorthalidone, respectively.
The inactive ingredients are ammonium chloride, colloidal silicon dioxide, croscarmellose sodium (Type A), magnesium stearate, microcrystalline cellulose, sodium lauryl sulfate, D&C yellow #10.
Clonidine Hydrochloride
Clonidine hydrochloride is an imidazoline derivative and exists as a mesomeric compound. The chemical name is 2-[(2,6-dichlorophenyl)imino]imidazoline monohydrochloride. The following are the structural formula, molecular formula and molecular weight:
 |
Clonidine hydrochloride is an odorless, bitter, white crystalline substance soluble in water and alcohol.
About BioAlliance Pharma
Dedicated to cancer and supportive care treatment with a focus on resistance targeting and orphan products, BioAlliance conceives and develops innovative products, for specialty markets especially in the hospital setting and for orphan or rare diseases.
Created in 1997 and introduced to the Euronext Paris market in 2005, BioAlliance Pharma’s ambition is to become a leading player in these fields by coupling innovation to patient needs. The company’s teams have the key competencies required to identify, develop and register drugs in Europe and the USA.

Teva Announces FDA Approval of Generic Adderall XR®

FEB 14 2013, Teva Pharmaceutical Industries Ltd. announced today that the U.S. Food and Drug Administration (FDA) has approved its Abbreviated New Drug Application (ANDA) for the generic version of Shire’s Adderall XR®Capsules, 5mg, 10mg, 15mg, 20mg, 25mg and 30 mg capsules for the treatment of attention deficit hyperactivity disorder. Adderall XR® had annual sales, including brand and generic sales, of approximately $2 billion in the United States, based on IMS sales data as of December 31, 2012.
Teva currently sells a generic version of Adderall XR® Capsules under a 2006 license and distribution agreement with Shire as part of a settlement of patent litigation between Shire and Teva’s subsidiary Barr Pharmaceuticals. Under the terms of the agreement, Teva has the right to be supplied product by Shire through April 1, 2014.
Adderall is a psychostimulant medication that contains amphetamine. It is used for the treatment of attention deficit hyperactivity disorder (ADHD) and narcolepsy.[1] Adderall is a combination of four amphetamine salts (racemic amphetamine aspartate monohydrate, racemic amphetamine sulfate, dextroamphetamine saccharide, and dextroamphetamine sulfate). It is a dopamine releasing agent, a norepinephrine releasing agent, and can be mildly serotonergic.[2] It is available in two formulations: IR (Instant Release) and XR (Extended Release). The immediate release formulation is indicated for use in attention deficit hyperactivity disorder (ADHD) and narcolepsy,[3] while the XR formulation is approved for use only with attention deficit hyperactivity disorder (ADHD).[2]
Important side effects of therapeutic dextroamphetamine include stunted growth in young people and occasionally a psychosis can occur at therapeutic doses during chronic therapy as a treatment emergent side effect.[4] When abused at high doses, the risk of experiencing side effects and the severity of side effects increases. Side effects may include sweating or shaking.
Like other stimulant drugs, such as methamphetamine and cocaine, Adderall directly affects the mesolimbic reward pathway in the brain. Amphetamine salt preparations are considered to have high abuse potential, and it is classified as Schedule II by the US DEA. With the Safe Streets and Communities Act in Canada, Adderall has been reclassified from Schedule III to Schedule I.[5]
- “Adderall”. The American Society of Health-System Pharmacists. http://www.drugs.com/monograph/adderall.html. Retrieved 3 April 2
- “Adderall XR prescribing information”. Shire US. March 2009. http://pi.shirecontent.com/PI/PDFs/AdderallXR_USA_ENG.PDF. Retrieved 2009-06-23.
- “ADDERALL (CII)” (PDF). Food and Drug Administration. February 2007. http://www.accessdata.fda.gov/drugsatfda_docs/label/2007/011522s040lbl.pdf. Retrieved 2009-06-23.
- Berman, SM.; Kuczenski, R.; McCracken, JT.; London, ED. (Feb 2009). “Potential adverse effects of amphetamine treatment on brain and behavior: a review.”. Mol Psychiatry 14 (2): 123–42. doi:10.1038/mp.2008.90. PMID 18698321.
- C-10
| Combination of | |
|---|---|
| Dextroamphetamine | Psychostimulant |
| Amphetamine | Psychostimulant |
| Clinical data | |
| Trade names | Adderall, Adderall XR |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a601234 |
| Licence data | US Daily Med:link |
| Pregnancy cat. | C (US) |
| Legal status | Schedule I (CA) Schedule II (US) |
| Dependence liability | High |
| Routes | (Medical) Oral, (Recreational) Oral, Insufflated, Intravenous |
| Identifiers | |
| CAS number | 51-64-9 300-62-9 |
Dermasciences-Diabetic foot ulcer Phase 3 trials of DSC-127, Nle3 A(l-7)
DSC-127
feb 2013
Dermasciences-Diabetic foot ulcer Phase 3 trials of DSC-127, Nle3 A(l-7)
University of Southern California
A method for treating a subject that has suffered combined (i) exposure to total body ionizing irradiation and (ii) burns, comprising administering to the subject an amount effective to treat the radiation effects and/or the burn of a peptide comprising at least 5 amino acids of a peptide of SEQ ID NO: l (Asp- Arg-Nle-Tyr-Ile-His-Pro), or a pharmaceutical salt thereof.
WO-2012106427,
A method for treating diabetic foot ulcers, comprising administering to a human patient suffering from a diabetic foot ulcer an amount of a peptide of at least 5 contiguous amino acids of Nle3 A(l-7) effective to treat the diabetic foot ulcer
DSC127 is an analog of a naturally occurring peptide, Angiotensin. It has been shown to increase keratinocyte proliferation, increase extracellular matrix production, and increase vascularization. Additionally, histological examination has shown thatDSC127 accelerated collagen deposition six-fold. All these help to accelerate dermal tissue repair. One potential method of action is the up-regulation of mesenchymal stem cells (MSCs) at the site of injury. MSCs originate in the human embryo and are considered to be multipotent — a type of stem cell that has not yet adopted a specific cellular phenotype. Such cells have the ability to differentiate into various types of cells found within the human body, including fibroblasts, adipose cells, muscle cells, bone cells, and skin cells.
The patented amino acid peptide DSC127 optimizes the well published wound healing capabilities of Angiotensin while removing all blood pressure effects of the compound.
Derma Sciences, under license from the University of Southern California (USC), is developing DSC-127 (USB-001), a topical formulation of an angiotensin analog NorLeu3-A(1-7) that recruits mesenchymal stem cells to the sites of tissue injury, for the potential treatment of diabetic foot ulcer
Wound Repair and Regeneration
Volume 20, Issue 4, pages 482–490, July-August 2012
http://onlinelibrary.wiley.com/doi/10.1111/j.1524-475X.2012.00804.x/full
NorLeu3-A(1–7) stimulation of diabetic foot ulcer healing: …………
Cerulean doses first patient in Phase 2 study of CRLX101 drug


Chemical structure of CRLX-101
(source: Svenson S, Wolfgang M, Hwang J, Ryan J, Eliasof S. J Control Release. 2011 Jul 15;153(1):49-55. Epub 2011 Mar 23.).
CRLX101 is a novel approach to cancer chemotherapy that is currently under investigation in human trials, and is an example of ananomedicine.
The agent represents a nanoparticle conjugate that consists of a drug delivery molecule, namely a cyclodextrin-based polymer (CDP) and an anti-cancer compound (camptothecin). It was developed by Dr. Mark E. Davis, professor of Chemical Engineering at theCalifornia Institute of Technology, and associates at Insert Therapeutics, Inc., now Calando Pharmaceuticals, Inc., hence the original name “IT-101”. Its novel delivery mode allows the agent, and thus the toxic anti-cancer component, to be preferentially accumulated in cancer tissue. In turn, toxic side effect are expected to be reduced. The technology was licensed by Calando and Caltech to Cerulean Pharma Inc., in June, 2009.
CRLX101 is a camptothececin-nanoparticle conjugate, which is a novel approach to cancer chemotherapy that is currently under investigation in human trials. CRLX101 represents a nanoparticle conjugate that consists of a drug delivery molecule, namely a cyclodextrin-based polymer (CDP) and an anti-cancer compound (camptothecin). It was developed by Dr. Mark E. Davis, professor of Chemical Engineering at the California Institute of Technology, and associates at Insert Therapeutics, Inc., now Calando Pharmaceuticals, Inc., hence the original name “IT-101”. Its novel delivery mode allows the agent, and thus the toxic anti-cancer component, to be preferentially accumulated in cancer tissue. In turn, toxic side effect are expected to be reduced. The technology was licensed by Calando and Caltech to Cerulean Pharma Inc., in June, 2009. (source: http://en.wikipedia.org/wiki/CRLX101).
Camptothecin (CPT) is a potent broad-spectrum anticancer agent that acts through inhibition of topoisomerase 1. Clinical development of CPT was unsuccessful due to poor drug solubility, insufficient in vivo stability of the active form, and toxicity. In order to address these issues, a polymeric nanoparticle comprised of cyclodextrin-poly(ethylene glycol) copolymer (CDP) conjugated to CPT (CRLX101) has been developed and Phase 2 clinical studies are ongoing. Camptothecin is conjugated to the polymer in its active form at 10-12 wt.% loading. CRLX101 self-assembles in solution into nanoparticles with an apparent solubility increase of >1000-fold as compared to the parent drug camptothecin.
Current developer: Calando Pharmaceuticals, Inc/Cerulean Pharma Inc.
Cerulean Pharma has dosed the first patient in a Phase 2 study of its investigational CRLX101 drug, designed for the treatment of extensive-stage small cell lung cancer (SCLC) patients sensitive to first-line platinum-based chemotherapy.
CRLX101, a tumor-targeted nanopharmaceutical, is a dual inhibitor of topoisomerase 1 and hypoxia-inducible factor-1a and releases camptothecin over an extended period of time.
The randomized study, which is being conducted at the University of Chicago School of Medicine and affiliated institutions, has enrolled 150-patient to compare the efficacy of CRLX101 with topotecan, a second-line therapy for relapsed SCLC.
The trial has co-primary endpoints of progression-free survival (PFS) and three-month PFS rate, claims the company.
During the company’s preclinical and Phase 1/2a clinical trial, CRLX101 has demonstrated significant anti-tumor activity.
Cerulean Pharma chief medical officer Edward Garmey said the company’s clinical experience with CRLX101 shows a benign safety profile.
“The standard of care in SCLC is not well tolerated, so if we can demonstrate an efficacy benefit versus standard of care, CRLX101 would have the added benefit of improved quality of life for these very sick patients,” Garmey added.
GSK files for EU approval of trametinib for melanoma

N-(3-{3-Cyclopropyl-5-[(2-fluoro-4-iodophenyl)amino]-6,8-dimethyl-2,4,7-trioxo-3,4,6,7-tetrahydropyrido[4,3-d]pyrimidin-1(2H)-yl}phenyl)acetamide
Trametinib (GSK1120212) is experimental cancer drug. It is a MEK inhibitor drug with anti-cancer activity.[1]
Trametinib had good results for V600E mutated metastatic melanoma in a phase III clinical trial.[2]
- Trametinib, NCI Drug Dictionary
- METRIC phase III study: Efficacy of trametinib (T), a potent and selective MEK inhibitor (MEKi), in progression-free survival (PFS) and overall survival (OS), compared with chemotherapy (C) in patients (pts) with BRAFV600E/K mutant advanced or metastatic melanoma (MM).
FEBRUARY 12, 2013
The European Medicines Agency has given GlaxoSmithKline’s melanoma drug an accelerated review.
The drug – a MEK inhibitor called trametinib – is seeking a European licence as both a monotherapy and in combination with GSK’s investigational BRAF inhibitor dabrafenib, for the treatment of patients with unresectable or metastatic melanoma with a BRAF V600 mutation.
The EMA’s Committee for Medicinal Products for Human Use (CHMP) has granted GSK’s request for accelerated assessment of this application, meaning it may be on the market within six months if approved.
The application includes data from a Phase III study of trametinib monotherapy compared to the established chemotherapy agents dacarbazine or paclitaxel monotherapy in patients with BRAF V600 mutation positive metastatic melanoma
It also includes data from a randomised Phase I/II study comparing dabrafenib monotherapy to combination therapy with dabrafenib and trametinib in patients with BRAF V600 mutation positive metastatic melanoma
“We initiated a randomised study very early in the development programme to test whether the novel-novel combination could circumvent resistance to single agent anti-BRAF therapy and are encouraged by the results from this Phase I/II trial,” said Dr Rafael Amado, head of oncology R&D at GSK.
“We are planning further regulatory submissions based on these data, in the US and other countries in the coming months,” he added.
An application that has been granted accelerated assessment will have a maximum review time of 150 days, although the CHMP can extend this if needs be.
In August 2012, GSK announced regulatory submissions for dabrafenib monotherapy as a treatment for BRAF V600 metastatic melanoma in Europe and the USA, as well as a US submission for trametinib monotherapy as a treatment for BRAF V600 metastatic melanoma.
Trametinib and dabrafenib are investigational medicines and their use as monotherapy or combination therapy is not approved anywhere in the world.
Roche’s Zelboraf (vemurafenib) is currently the only licensed drug to treat BRAF positive melanoma patients, and is the first drug to increase overall survival in this patient population.
GSK is currently conducting a head-head Phase III trial against Zelboraf, with both of its drugs, results of which are expected next year. If approved GSK’s drugs will also be up against Bristol-Myers Squibb’s vaccine Yervoy, which works as a cancer vaccine.
Yervoy and Zelboraf are both expected to bring in peak annual sales of around $1 – $2 billion, making the new melanoma market a potentially lucrative venture for pharma.
OncoGenex Announces Plans for the Initiation of the Borealis-2 Clinical Trial Evaluating OGX-427 in Combination with Second-Line Therapy for Bladder Cancer
An antisense oligonucleotide of the form: GGGACGCGGCGCTCGTCAT

OGX-427 is a second generation antisense drug which, in preclinical experiments, inhibits production of Heat Shock Protein 27 (Hsp27), a cell survival protein found at elevated levels in many human cancers. The development program for OGX-427 aims to demonstrate inhibition of Hsp27 can lead to improved prognosis and treatment outcomes for cancer patients.
OncoGenex Announces Plans for the Initiation of the Borealis-2 Clinical Trial Evaluating OGX-427 in Combination with Second-Line Therapy for Bladder Cancer
Feb. 12, 2013
OncoGenex Pharmaceuticals, Inc. today announced plans for the initiation of the Borealis-2 clinical trial, an investigator-sponsored, randomized, controlled Phase 2 study evaluating OGX-427 in patients with advanced or metastatic bladder cancer who have disease progression following initial platinum-based chemotherapy treatment. The trial, which is the fourth Phase 2 study of OGX-427 in a genitourinary (GU) cancer, will investigate if combining OGX-427 with docetaxel, a standard option in salvage treatment for metastatic bladder cancer, improves survival compared to docetaxel alone.
“Bladder cancer is often sensitive to chemotherapy in the first-line setting, but, when patients relapse, resistance to chemotherapy is frequent,” stated Jonathan Rosenberg MD, Associate Physician, Memorial Sloan-Kettering Cancer Center and one of the primary investigators on the study. “This trial will evaluate the potential of OGX-427 to work synergistically with second- or third-line chemotherapy to overcome treatment resistance and prolong survival in patients with advanced bladder cancer.
“Limited options exist for both the first- and second-line treatment of advanced bladder cancer. Currently, first-line platinum-based chemotherapy regimens result in a median overall survival of approximately 12-15 months. Docetaxel is commonly used in second-line treatment, with a reported median overall survival of approximately six months. Given acquired treatment resistance and these short survival times, there continues to be a high unmet need for additional therapeutic options for this patient population.
OGX-427 is designed to inhibit Heat Shock Protein 27 (HSP27), a cell-survival protein found at elevated levels in many human cancers including prostate, bladder, breast and non-small cell lung cancer. Overexpression of Hsp27 is thought to be an important factor leading to the development of treatment resistance and is associated with negative clinical outcomes in patients with various tumor types.
“The launch of Borealis-2 marks OncoGenex’ continued commitment to expanding the OGX-427 clinical development program to better understand treatment resistance in GU cancers,” said Scott Cormack, President and Chief Executive Officer of OncoGenex. “Given the growing incidence of bladder cancer due to an aging population, we believe there is an urgent need to identify new strategies to address treatment resistance and potentially improve outcomes in this patient population.”
Borealis-2 will be the second randomized, controlled clinical trial of OGX-427 in advanced bladder cancer. The Borealis-1 clinical trial is the OncoGenex-sponsored, randomized, placebo-controlled Phase 2 study designed to evaluate a potential survival benefit, safety and tolerability of combining OGX-427 with gemcitabine and cisplatin in the first-line treatment of patients with advanced bladder cancer. If either Borealis trial shows a survival advantage, OncoGenex plans to initiate conversations with the Food and Drug Administration about the possibility of a Phase 3 study of OGX-427 in bladder cancer as part of the ORCA program.
About Borealis-2
The Borealis-2 clinical trial will randomize approximately 200 patients to receive either OGX-427 plus docetaxel treatment or docetaxel treatment alone. Patients may also continue weekly OGX-427 infusions as maintenance treatment until disease progression or unacceptable toxicity if they complete all 10 planned cycles of docetaxel or are discontinued from docetaxel due to docetaxel toxicity. The primary objective will be overall survival, with secondary objectives evaluating safety, tolerability, tumor response rates and the effect of therapy on Hsp27 levels and circulating tumor cells.Borealis-2 will be conducted at approximately 30 sites in the U.S. and is being sponsored by the Hoosier Oncology Group. Dr. Noah Hahn from the Indiana University Simon Cancer Center, Dr. Toni Choueiri from the Dana-Farber Cancer Institute and Dr. Jonathan Rosenberg from Memorial Sloan-Kettering Cancer Center will serve as the primary investigators on the study.
ABOUT ORCA
The “ORCA” (Overcoming Resistance in CAncer) program encompasses the on-going studies of OGX-427 aiming to demonstrate that inhibition of Hsp27 can lead to improved prognosis and treatment outcomes for cancer patients. Phase 2 clinical trials are underway in prostate and bladder cancers, with additional studies expected to initiate this year. For more information on OGX-427 and ORCA, please visit http://www.oncogenex.com.
