
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

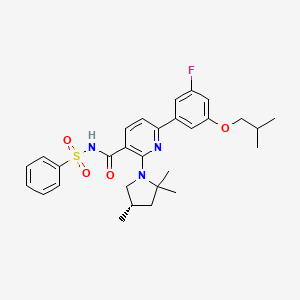
OLACAFTOR, VX 440
CAS 1897384-89-2
| Molecular Formula: | C29H34FN3O4S |
|---|---|
| Molecular Weight: | 539.666 g/mol |
CFTR corrector; UNII-RZ7027HK8F; RZ7027HK8F;
Target-based Actions, CFTR modulator
Indications, Cystic fibrosis
Olacaftor (VX-440, VX440) is a next-generation CFTR corrector, shows the potential to enhance the amount of CFTR protein at the cell’s surface and for treatment of cystic fibrosis..
- Originator Vertex Pharmaceuticals
- Class Pyridines; Pyrrolidines
- Mechanism of Action Cystic fibrosis transmembrane conductance regulator stimulants
- Phase II Cystic fibrosis
- 01 Jun 2018 Chemical structure information added
- 01 Aug 2017 Vertex Pharmaceuticals completes a phase II trial in Cystic fibrosis (In adolescents, In adults, In the elderly, Combination therapy) in USA, Australia, Austria, Belgium, Canada, Denmark, Germany, Italy, Spain, Netherlands and United Kingdom (PO) (NCT02951182) (EudraCT2016-000454-36)
- 18 Jul 2017 Efficacy and events data from a phase II trial in Cystic fibrosis released by Vertex Pharmaceuticals
PATENT
WO2016057572
PATENT
PATENT
WO-2019028228
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019028228&tab=PCTDESCRIPTION&maxRec=1000
Processes for preparing (S)-2,2,4-trimethylpyrrolidine and its salts, particularly hydrochloride comprising the reaction of 2,2,6,6-tetramethyl-piperidin-4-one with chloroform and a base (sodium hydroxide), followed by reaction with an acid (hydrochloric acid), hydrogenation, reduction and salt synthesis is claimed. Also claimed is a process for the preparation of an intermediate of (S)-2,2,4-trimethylpyrrolidine hydrochloride. The compound is useful as an intermediate for the synthesis of CFTR modulators, useful for treating cystic fibrosis.
(5)-2,2,4-trimethylpyrrolidine free base and salt forms thereof, (R)-2,2,4-trimethylpyrrolidine free base and salt forms thereof, (,S)-3,5,5-trimethylpyrrolidine-2-one, (R)-3,5,5-trimethylpyrrolidine-2-one, and 5,5-dimethyl-3-methylenepyrrolidin-2-one are useful molecules that can be used in the synthesis of pharmaceutically active molecules, such as modulators of CFTR activity, for example those disclosed in PCT Publication Nos. WO 2016/057572, WO 2018/064632, and WO 2018/107100, including the following molecules, which are being investigated in clinical trials for the treatment of cystic fibrosis:
[0003] There remains, however, a need for more efficient, convenient, and/or economical processes for the preparation of these molecules.
[0004] Disclosed herein are processes for preparing 5,5-dimethyl-3-methylenepyrrolidin-2-one, (,S)-3,5,5-trimethylpyrrolidine-2-one, (R)-3,5,5-trimethylpyrrolidine-2-one, (,S)-2,2,4-trimethylpyrrolidine, and (R)-2,2,4-trimethylpyrrolidine, and their salt forms:
trimethylpyrrolidine-2-one)); ((R)-3,5,5-trimethylpyrrolidine-2-one));
((,S)-2,2,4-trimethylpyrrolidine) ;and
Scheme 1. Synthesis of (S)-2,2,4-trimethylpyrrolidine
(2) (3) (4S) (1 S)
Scheme 2. Synthesis of (R)-2,2,4-trimethylpyrrolidine
(2) (3) (4R) (1 R)
Scheme 3. Synthesis of 5,5-dimethyl-3-methylenepyrrolidin-2-one
3 C
EXAMPLES
Example 1. Reaction (a) and (b): Synthesis of 5,5-dimethyl-3-methylenepyrrolidin- 2-one
(2) (3) C (3)
Example 1A:
[0055] 2,2,6,6-tetramethylpiperidin-4-one (50.00 g, 305.983 mmol, 1.000 equiv), tributylmethylammonium chloride (2.89 g, 3.0 mL, 9.179 mmol, 0.030 equiv), chloroform (63.92 g, 43.2 mL, 535.470 mmol, 1.750 equiv), and DCM (dichloromethane) (100.0 mL, 2.00 vol) were charged to a 1000 mL three-neck round bottom flask equipped with an overhead stirrer. The reaction mixture was stirred at 300 rpm, and 50 wt% NaOH (195.81 g, 133.2 mL, 2,447.863 mmol, 8.000 equiv) was added dropwise (via addition funnel) over 1.5 h while maintaining the temperature below 25 °C with intermittent ice/acetone bath. The reaction mixture was stirred at 500 rpm for 18 h, and monitored by GC (3% unreacted piperidinone after 18 h). The suspension was diluted with DCM (100.0 mL, 2.00 vol) and H2O (300.0 mL, 6.00 vol), and the phases were separated. The aqueous phase was extracted with DCM (100.0 mL, 2.00 vol). The organic phases were combined and 3 M hydrochloric acid (16.73 g, 153.0 mL, 458.974 mmol, 1.500 equiv) was added. The mixture was stirred at 500 rpm for 2 h. The conversion was complete after approximately 1 h. The aqueous phase was saturated with NaCl, H2O (100.0 mL, 2.00 vol) was added to help reduce the emulsion, and the phases were separated. The aqueous phase was extracted with DCM (100.0 mL, 2.00 vol) twice. H2O (100.0 mL, 2.00 vol) was added to help with emulsion separation. The organic phases were combined, dried (MgS04), and
concentrated to afford 32.6 g (85%) of crude Compound (3) as a pale orange clumpy solid. The crude was recrystallized from hot (90°C) iPrOAc (isopropyl acetate) (71.7 mL, 2.2 vol. of crude), cooled to 80 °C, and -50 mg of crystalline Compound (3) was added for seeding. Crystallization started at 77 °C, the mixture was slowly cooled to ambient temperature, and aged for 2 h. The solid was collected by filtration, washed with 50/50 iPrOAc/heptane (20.0 mL, 0.40 vol) twice, and dried overnight in the vacuum oven at 40 °C to afford the desired product (23.70 g, 189.345 mmol, 62% yield) as a white sand colored crystalline solid. ¾ MR (400 MHz, CDCh, 7.26 ppm) δ 7.33 (bs, 1H), 5.96-5.95 (m, 1H), 5.31-5.30 (m, 1H), 2.6 (t, J= 2.5 Hz, 2H), 1.29 (s, 6H).
Synthesis IB:
[0056] i. Under a nitrogen atmosphere, 2,2,6,6-tetramethylpiperidin-4-one (257.4 kg, 1658.0 mol, 1.00 eq.), tri-butyl methyl ammonium chloride (14.86 kg, 63.0 mol, 0.038 eq.), chloroform (346.5 kg, 2901.5 mol, 1.75 eq.) and DCM (683.3 kg) were added to a 500 L enamel reactor. The reaction was stirred at 85 rpm and cooled to 15~17°C. The solution of 50wt% sodium hydroxide (1061.4 kg, 13264.0 mol, 8.00 eq.) was added dropwise over 40 h while maintaining the temperature between 15~25°C. The reaction mixture was stirred and monitored by GC.
ii. The suspension was diluted with DCM (683.3 kg) and water (1544.4 kg). The organic phase was separated. The aqueous phase was extracted with DCM (683.3 kg). The organic phases were combined, cooled to 10°C and then 3 M hydrochloric acid (867.8 kg, 2559.0 mol, 1.5 eq.) was added. The mixture was stirred at 10-15 °C for 2 h. The organic phase was separated. The aqueous phase was extracted with DCM (683.3 kg x 2). The organic phases were combined, dried over Na2S04 (145.0 kg) for 6 h. The solid was filtered off and washed with DCM (120.0 kg). The filtrate was stirred with active charcoal (55 kg) for 6 h. The resulting mixture was filtered and the filtrate was concentrated under reduced pressure (30~40°C, -O. lMPa). Then isopropyl acetate (338 kg) was added and the mixture was heated to 87-91°C, stirred for 1 h. Then the solution was cooled to 15 °C in 18 h and stirred for 1 h at 15 °C. The solid was collected by filtration, washed with 50% isopropyl acetate/hexane (80.0 kg x 2) and dried overnight in the vacuum oven at 50 °C to afford 5,5-dimethyl-3-methylenepyrrolidin-2-one as an off white solid, 55% yield.
Example 2. Reaction (c): Synthesis of (S)-3,5,5-trimethyl-pyrrolidin-2-one from 5,5-dimethyl-3-methylenepyrrolidin-2-one
(3) (4S)
Example 2A: Use of Rh Catalyst
[0057] Step 1 : Preparation of Rh Catalyst Formation: In a 3 L Schlenk flask, 1.0 L of tetrahydrofuran (THF) was degassed with an argon stream. Mandyphos Ligand SL-M004-1 (1.89 g) and [Rh(nbd)Cl]2 (98%, 0.35 g) (chloronorbornadiene rhodium(I) dimer) were added. The resulting orange catalyst solution was stirred for 30 min at room temperature to form a catalyst solution.
[0058] Step 2: A 50 L stainless steel autoclave was charged with 5,5-dimethyl-3-■methylenepyrrolidin-2-one (6.0 kg, Compound (3)) and THF (29 L). The autoclave was
sealed and the resulting suspension was flushed with nitrogen (3 cycles at 10 bar), and then released of pressure. Next the catalyst solution from Step 1 was added. The autoclave was flushed with nitrogen without stirring (3 cycles at 5 bar) and hydrogen (3 cycles at 5 bar). The pressure was set to 5 bar and a 50 L reservoir was connected. After 1.5 h with stirring at 1000 rpm and no hydrogen uptake the reactor was flushed again with nitrogen (3 cycles at 10 bar) with stirring and additional catalyst solution was added. The autoclave was again flushed to hydrogen with the above described procedure (3 x 5 bar N2, 3 x 5 bar H2) and adjusted to 5 bar. After 2 h, the pressure was released, the autoclave was flushed with nitrogen (3 cycles at 5 bar) and the product solution was discharged into a 60 L inline barrel. The autoclave was charged again with THF (5 L) and stirred with 1200 rpm for 5 min. The wash solution was added to the reaction mixture.
[0059] Step 3 : The combined solutions were transferred into a 60 L reactor. The inline barrel was washed with 1 L THF which was also added into the reactor. 20 L THF were removed by evaporation at 170 mbar and 40°C. 15 L heptane were added. The distillation was continued and the removed solvent was continuously replaced by heptane until the THF content in the residue was 1% w/w (determined by NMR). The reaction mixture was heated to 89°C (turbid solution) and slowly cooled down again (ramp: 14°C/h). Several heating and cooling cycles around 55 to 65°C were made. The off-white suspension was transferred to a stirred pressure filter and filtered (ECTFE-pad, d = 414 mm, 60 my, Filtration time = 5 min). 10 L of the mother liquor was transferred back into the reactor to wash the crystals from the reactor walls and the obtained slurry was also added to the filter. The collected solid was washed with 2 x 2.5 1 heptane, discharged and let dry on the rotovap at 40°C and 4 mbar to obtain the product, (S)-3,5,5-trimethyl-pyrrolidin-2-one; 5.48 Kg (91%), 98.0% ee.
Synthesis 2B: Use of Ru Catalyst
[0060] The reaction was performed in a similar manner as described above in Example 2A except the use of a Ru catalyst instead of a Rh catalyst.
[0061] Compound (3) (300 g) was dissolved in THF (2640 g, 10 Vol) in a vessel. In a separate vessel, a solution of [RuCl(p-cymene){(R)-segphos}]Cl (0.439g, 0.0002 eq) in THF (660 g, 2.5 Vol) was prepared. The solutions were premixed in situ and passed
through a Plug-flow reactor (PFR). The flow rate for the Compound (3) solution was at 1.555 mL/min and the Ru catalyst solution was at 0.287 mL/min. Residence time in the PFR was 4 hours at 30 °C, with hydrogen pressure of 4.5 MPa. After completion of reaction, the TFIF solvent was distilled off to give a crude residue. Heptane (1026 g, 5 vol) was added and the resulting mixture was heated to 90 °C. The mixture was seeded with 0.001 eq. of Compound 4S seeds. The mixture was cooled to -15 °C at 20 °C/h. After cooling, heptane (410 g, 2 vol) was added and the solid product was recovered by filtration. The resulting product was dried in a vacuum oven at 35 °C to give (S)-3,5,5-trimethyl-pyrrolidin-2-one (281.77 g, 98.2 % ee, 92 % yield).
Example 2C: Analytical Measurements
[0062] Analytical chiral HPLC method for the determination of the conversion, chemoselectivity and enantiomeric excess of the products form Example 2A and 2B was made under the following conditions: Instrument: Agilent Chemstation 1100; Column: Phenomenex Lux 5u Cellulose— 2, 4.6 mm x 250 mm x 5 um, LHS6247; Solvent:
Heptane/iPrOH (90: 10); Flow: 1.0 ml/min; Detection: UV (210 nm); Temperature: 25°C; Sample concentration: 30 μΐ of reaction solution evaporated, dissolved in 1 mL;
heptane/iPrOH (80/20); Injection volume: 10.0
Run time 20 min; Retention times: 5,5–dimethyl-3-methylenepyrrolidin-2-one: 13.8 min, (,S)-3,5,5-trimethyl-pynOlidin-2-one: 10.6 min, and (R)-3,5,5-trimethyl-pyrrolidin-2-one: 12.4 min.
Example 3: Alternate Synthesis of (S)-3,5,5-trimethyl-pyrrolidin-2-one from 5,5-dimethyl-3-methylenepyrrolidin-2-one
Ru(Me-allyl)2(C0D)2BF4
1 eq HBF4 Et20
5 bar H2 at 45°C
[0063] Mandyphos (0.00479 mmol, 0.12 eq) was weighed into a GC vial. In a separate vial, Ru(Me-allyl)2(COD) (16.87 mg, 0.0528 mmol) was weighed and dissolved in DCM (1328 \iL). In another vial HBF4 Et20 (6.6 μΐ,) and BF3 Et20 (2.0 μΐ,) were dissolved in DCM (240 μΐ.). To the GC vial containing the ligand was added, under a flow of argon, the Ru(Me-allyl)2(COD) solution (100 μΐ,; 0.00399 mmol, O. leq) and the HBF4 Et20 / BF3 -Et20 solution (20 μΐ^ 1 eq HBF4 Et20 and catalytic BF3 Et20). The resulting mixtures were stirred under a flow of argon for 30 minutes. 5,5-dimethyl-3-methylenepyrrolidin-2-one (5 mg, 0.0399 mmol) in EtOH (1 mL) was added. The vials were placed in the hydrogenation apparatus. The apparatus was flushed with H2 (3 χ) and charged with 5 bar H2. After standing for 45 minutes, the apparatus was placed in an oil bath at temperature of 45°C. The reaction mixtures were stirred overnight under H2. 200 μΙ_, of the reaction mixture was diluted with MeOH (800 μΐ.) and analyzed for conversion and ee. 1H MR (400 MHz, Chloroform-d) δ 6.39 (s, 1H), 2.62 (ddq, J = 9.9, 8.6, 7.1 Hz, 1H), 2.17 (ddd, J = 12.4, 8.6, 0.8 Hz, 1H), 1.56 (dd, J = 12.5, 9.9 Hz, 1H), 1.31 (s, 3H), 1.25 (s, 3H), 1.20 (d, J = 7.1 Hz, 3H).
IPC analytical method for Asymmetric Hydrogenation
(3) (4S) (4R)
Example 4. Synthesis of (S)-2,2,4-trimethylpyrrolidine hydrochloride from (S)-3,5,5-trimethyl-pyrrolidin-2-one
(4S) (1S)HCI
Example 4A:
[0064] Anhydrous THF (100 ml) was charged to a dry 750 ml reactor and the jacket temperature was set to 50° C. Once the vessel contents were at 50° C, LiAlH4pellets (10 g, 263 mmol, 1.34 eq.) were added. The mixture was stirred for 10 minutes, then a solution of (4S) (25 g, 197 mmol) in anhydrous THF (100 ml) was added dropwise over 45 minutes, maintaining the temperature between 50-60° C. Once the addition was complete the jacket temperature was increased to 68° C and the reaction was stirred for 18.5 hrs. The reaction mixture was cooled to 30° C then saturated sodium sulfate solution (20.9 ml) was added dropwise over 30 minutes, keeping the temperature below 40° C. Vigorous evolution of hydrogen was observed and the reaction mixture thickened but remained mixable. The mixture thinned towards the end of the addition. The mixture was cooled to 20° C, diluted with iPrOAc (100 ml) and stirred for an additional 10 minutes. The suspension was then drained and collected through the lower outlet valve, washing through with additional iPrOAc (50 ml). The collected suspension was filtered through a Celite pad on a sintered glass funnel under suction and washed with iPrOAc (2×50 ml).
[0065] The filtrate was transferred back to the cleaned reactor and cooled to 0° C under nitrogen. 4M HCI in dioxane (49.1 ml, 197 mmol, leq.) was then added dropwise over 15 minutes, maintaining the temperature below 20°C. A white precipitate formed. The reactor was then reconfigured for distillation, the jacket temperature was increased to 100 °C, and distillation of solvent was carried out. Additional z-PrOAc (100 mL) was added during concentration, after >100 mL distillate had been collected. Distillation was continued until -250 mL total distillate was collected, then a Dean-Stark trap was attached and reflux continued for 1 hour. No water was observed to collect. The reaction mixture was cooled to 20 °C and filtered under suction under nitrogen. The filtered solid was washed with i-PrOAc (100 mL), dried under suction in nitrogen, then transferred to a glass dish and dried in a vacuum oven at 40 °C with a nitrogen bleed. Compound (1S)»HC1 was obtained as a white solid (24.2g, 82%).
Synthesis 4B:
[0066] To a glass lined 120 L reactor was charged LiAlH4 pellets (2.5 kg 66 mol, 1.2 equiv.) and dry THF (60 L) and warmed to 30 °C. To the resulting suspension was charged (¾)-3,5,5-trimethylpyrrolidin-2-one (7.0 kg, 54 mol) in THF (25 L) over 2 hours while maintaining the reaction temperature at 30 to 40 °C. After complete addition, the reaction temperature was increased to 60 – 63 °C and maintained overnight. The reaction mixture was cooled to 22 °C and sampled to check for completion, then cautiously quenched with the addition of EtOAc (1.0 L, 10 moles, 0.16 eq) followed by a mixture of THF (3.4 L) and water (2.5 kg, 2.0 eq) then followed by a mixture of water (1.75 kg) with 50 % aqueous sodium hydroxide (750 g, 2 eq water with 1.4 eq sodium hydroxide relative to aluminum), followed by 7.5 L water (6 eq “Fieser” quench). After the addition was completed, the reaction mixture was cooled to room temperature, and the solid was removed by filtration and washed with THF (3 x 25 L). The filtrate and washings were combined and treated with 5.0 L (58 moles) of aqueous 37% HC1 (1.05 equiv.) while maintaining the temperature below 30°C. The resultant solution was concentrated by vacuum distillation to a slurry in two equal part lots on the 20 L Buchi evaporator.
Isopropanol (8 L) was charged and the solution reconcentrated to near dryness by vacuum distillation. Isopropanol (4 L) was added and the product slurried by warming to about 50 °C. Distillation from Isopropanol continued until water content by KF is < 0.1 %. Methyl tertbutyl ether (6 L) was added and the slurry cooled to 2-5 °C. The product was collected by filtration and rinsed with 12 L methyl tert-butyl ether and pulled dry with a strong nitrogen flow and further dried in a vacuum oven (55 °C/300 torr/N2 bleed) to afford (S)-2,2,4-trimethylpyrrolidine»HCl ((1S HC1) as a white, crystalline solid (6.21 kg, 75% yield). ¾ NMR (400 MHz, DMSO-^6) δ 9.34 (s, 2H), 3.33 (dd, J= 11.4, 8.4 Hz, 1H), 2.75 (dd, J= 11.4, 8.6 Hz, 1H), 2.50 – 2.39 (m, 1H), 1.97 (dd, 7= 12.7, 7.7 Hz, 1H), 1.42 (s, 3H), 1.38 (dd, 7= 12.8, 10.1 Hz, 1H), 1.31 (s, 3H), 1.05 (d, 7= 6.6 Hz, , 3H).
Synthesis 4C:
[0067] With efficient mechanical stirring, a suspension of LiAlH4 pellets (100 g 2.65 mol; 1.35 eq.) in THF (1 L; 4 vol. eq.) warmed at a temperature from 20 °C – 36 °C (heat of mixing). A solution of (¾)-3,5,5-trimethylpyrrolidin-2-one (250 g; 1.97 mol) in THF (1 L; 4 vol. eq.) was added to the suspension over 30 min. while allowing the reaction temperature to rise to -60 °C. The reaction temperature was increased to near reflux (-68 °C) and maintained for about 16 h. The reaction mixture was cooled to below 40 °C and cautiously quenched with drop-wise addition of a saturated aqueous solution of Na2S04 (209 mL) over 2 h. After the addition was completed, the reaction mixture was cooled to ambient temperature, diluted with /-PrOAc (1 L), and mixed thoroughly. The solid was removed by filtration (Celite pad) and washed with /‘-PrOAc (2 x 500 mL). With external cooling and N2 blanket, the filtrate and washings were combined and treated with drop-wise addition of anhydrous 4 M HC1 in dioxane (492 mL; 2.95 mol; 1 equiv.) while maintaining the temperature below 20 °C. After the addition was completed (20 min), the resultant suspension was concentrated by heating at reflux (74 – 85 °C) and removing the distillate. The suspension was backfilled with /‘-PrOAc (1 L) during concentration. After about 2.5 L of distillate was collected, a Dean-Stark trap was attached and any residual water was azeotropically removed. The suspension was cooled to below 30 °C when the solid was collected by filtration under a N2 blanket. The solid is dried under N2 suction and further dried in a vacuum oven (55 °C/300 torr/N2 bleed) to afford 261 g (89% yield) of (S 2,2,4-trimethylpyrrolidine»HCl ((1S HC1) as a white, crystalline solid. ¾ NMR (400 MHz, DMSO-^6) δ 9.34 (s, 2H), 3.33 (dd, J = 11 A, 8.4 Hz, 1H), 2.75 (dd, J= 11.4, 8.6 Hz, 1H), 2.50 – 2.39 (m, 1H), 1.97 (dd, J= 12.7, 7.7 Hz, 1H), 1.42 (s, 3H), 1.38 (dd, J = 12.8, 10.1 Hz, 1H), 1.31 (s, 3H), 1.05 (d, J= 6.6 Hz, 3H). ¾ MR (400 MHz, CDCh) δ 9.55 (d, J= 44.9 Hz, 2H), 3.52 (ddt, J= 12.1, 8.7, 4.3 Hz, 1H), 2.94 (dq, J= 11.9, 5.9 Hz, 1H), 2.70 – 2.51 (m, 1H), 2.02 (dd, J= 13.0, 7.5 Hz, 1H), 1.62 (s, 3H), 1.58 – 1.47 (m, 4H), 1.15 (d, J= 6.7 Hz, 3H).
Synthesis 4D:
[0068] A 1L four-neck round bottom flask was degassed three times. A 2M solution of LiAlHun THF (100 mL) was charged via cannula transfer. (¾)-3,5,5-trimethylpyrrolidin-2-one (19.0 g) in THF (150 mL) was added dropwise via an addition funnel over 1.5 hours at 50-60 °C, washing in with THF (19 mL). Upon completion of the addition, the reaction was stirred at 60 °C for 8 hours and allowed to cool to room temperature overnight. GC analysis showed <1% starting material remained. Deionized water (7.6 mL) was added slowly to the reaction flask at 10-15 °C, followed by 15% potassium hydroxide (7.6 mL). Isopropyl acetate (76 mL) was added, the mixture was stirred for 15 minutes and filtered, washing through with isopropyl acetate (76 mL). The filtrate was charged to a clean and dry 500 mL four neck round bottom flask and cooled to 0-5 °C. 36% Hydrochloric acid (15.1 g, 1.0 eq.) was added keeping the temperature below 20 °C. Distillation of the solvent, backfilling with isopropyl acetate (190 mL), was carried out to leave a residual volume of -85 mL. Karl Fischer analysis = 0.11% w/w H2O. MTBE (methyl tertiary butyl ether) (19 mL) was added at 20-30 °C and the solids were filtered off under nitrogen at 15-20 °C, washing with isopropyl acetate (25 mL) and drying under vacuum at 40-45 °C to give crude (,S)-2,2,4-trimethylpyrrolidine hydrochloride as a white crystalline solid (17.4 g, 78% yield). GC purity = 99.5%. Water content = 0.20% w/w. Chiral GC gave an ee of 99.0% (S). Ruthenium content = 0.004 ppm. Lithium content = 0.07 ppm. A portion of the dried crude ,S)-2,2,4-trimethylpyrrolidine hydrochloride (14.3g) was charged to a clean and dry 250 mL four-neck round bottom flask with isopropanol (14.3 mL) and the mixture held at 80-85 °C (reflux) for 1 hour to give a clear solution. The solution was allowed to cool to 50 °C (solids precipitated on cooling) then MTBE (43 mL) was added and the suspension held at 50-55 °C (reflux) for 3 hours. The solids were filtered off at 10 °C, washing with MTBE (14 mL) and dried under vacuum at 40 °C to give recrystallised (S)- 2.2.4- trimethylpyrrolidine hydrochloride ((1S)»HC1) as a white crystallised solid (13.5 g, 94% yield on recrystallisation, 73% yield). GC purity = 99.9%. Water content = 0.11% w/w. 99.6% ee (Chiral GC) (S). Ruthenium content = 0.001 ppm. Lithium content = 0.02 ppm.
Synthesis 4E:
[0069] A reactor was charged with lithium aluminum hydride (LAH) (1.20 equiv.) and 2-MeTHF (2-methyltetrahydrofuran) (4.0 vol), and heated to internal temperature of 60 °C while stirring to disperse the LAH. A solution of (¾)-3,5,5-trimethylpyrrolidin-2-one (1.0 equiv) in 2-MeTHF (6.0 vol) was prepared and stirred at 25 °C to fully dissolve the (S)- 3.5.5- trimethylpyrrolidin-2-one. The (¾)-3,5,5-trimethylpyrrolidin-2-one solution was added slowly to the reactor while keeping the off-gassing manageable, followed by rinsing the addition funnel with 2-MeTHF (1.0 vol) and adding it to the reactor. The reaction was stirred at an internal temperature of 60 ± 5 °C for no longer than 6 h. The internal temperature was set to 5 ± 5 °C and the agitation rate was increased. A solution of water (1.35 equiv.) in 2-MeTHF (4.0v) was prepared and added slowly to the reactor while the internal temperature was maintained at or below 25 °C. Additional water (1.35 equiv.) was charged slowly to the reactor while the internal temperature was maintained at or below 25 °C. Potassium hydroxide (0.16 equiv.) in water (0.40 vol) was added to the reactor over no less than 20 min while the temperature was maintained at or below 25 °C. The resulting solids were removed by filtration, and the reactor and cake were washed with 2-MeTHF (2 x 2.5 vol). The filtrate was transferred back to a jacketed vessel, agitated, and the temperature was adjusted to 15 ± 5 °C. Concentrated aqueous HC1 (35-37%, 1.05 equiv.) was added slowly to the filtrate while maintaining the temperature at or below 25 °C and was stirred no less than 30 min. Vacuum was applied and the solution was distilled down to a total of 4.0 volumes while maintaining the internal temperature at or below 55 °C, then 2-MeTHF (6.00 vol) was added to the vessel. The distillation was repeated until Karl Fischer analysis (KF) < 0.20% w/w H2O. Isopropanol was added (3.00 vol), and the temperature was adjusted to 70 °C (65 – 75 °C) to achieve a homogenous solution, and stirred for no less than 30 minutes at 70 °C. The solution was cooled to 50 °C (47 – 53 °C) over 1 hour and stirred for no less than 1 h, while the temperature was maintained at 50°C (47 – 53 °C). The resulting slurry was cooled to -10 °C (-15 to -5°C) linearly over no less than 12 h. The slurry was stirred at -10 °C for no less than 2 h. The solids were isolated via filtration or centrifugation and were washed with a solution of 2-MeTHF (2.25 vol) and IPA (isopropanol) (0.75 vol). The solids were dried under vacuum at 45 ± 5 °C for not less than 6 h to yield (,S)-2,2,4-trimethylpyrrolidine hydrochloride ((1S)»HC1).
Example 5: Phase Transfer Catalyst (PTC) Screens for the Synthesis of 5,5-dimethyl-3-methylenepyrrolidin-2-one
[0070] Various PTCs were tested as described below:
[0071] 2,2,6,6-tetramethylpiperidin-4-one (500.0 mg, 3.06 mmol, 1.0 eq.), PTC (0.05 eq.), and chloroform (0.64 g, 0.4 mL, 5.36 mmol, 1.75 eq.) were charged into a vial equipped with a magnetic stir bar. The vial was cooled in an ice bath and a solution of 50 wt% sodium hydroxide (0.98 g, 24.48 mmol, 8.0 eq.) was added dropwise over 2 min. The reaction mixture was stirred until completion as assessed by GC analysis. The reaction mixture was diluted with DCM (2.0 mL, 4.0v) and H2O (3.0 mL, 6.0v). The phases were separated and the aqueous phase was extracted with DCM (1.0 mL, 2.0v). The organic
phases were combined and 2 M hydrochloric acid (0.17 g, 2.3 mL, 4.59 mmol, 1.5 eq.) was added. The reaction mixture was stirred until completion and assessed by
HPLC. The aqueous phase was saturated with NaCl and the phases were separated. The aqueous phase was extracted with DCM (1.0 mL, 2.0v) twice, the organic phases were combined, and 50 mg of biphenyl in 2 mL of MeCN was added as an internal HPLC standard. Solution yield was assessed by HPLC. The reaction results are summarized in the following table:
Example 6: Solvent Screens for the Synthesis of 5,5-dimethyl-3-methylenepyrrolidin-2-one
[0072] Various solvents and amounts were tested as described below:
[0073] 2,2,6,6-tetramethylpiperidin-4-one (500.0 mg, 3.06 mmol, 1.0 eq. (“starting material”)), tetrabutylammonium hydroxide (0.12 g, 0.153 mmol, 0.050 eq), chloroform (0.64 g, 0.4 mL, 5.36 mmol, 1.75 eq.), and solvent (2v or 4v, as shown below) were charged into a vial equipped with a magnetic stir bar. The vial was cooled in an ice bath and a solution of 50 wt% sodium hydroxide (0.98 g, 24.48 mmol, 8.0 eq.) was added drop wise over 2 min. The reaction mixture was stirred until completion and assessed by GC analysis. The reaction mixture was diluted with DCM (2.0 mL, 4.0v) and H2O (3.0 mL, 6.0v). The phases were separated and the aqueous phase was extracted with DCM (1.0 mL, 2.0v). The organic phases were combined and 2 M hydrochloric acid (0.17 g, 2.3 mL, 4.59 mmol, 1.5 eq.) was added. The reaction mixture was stirred until completion, assessed by HPLC. The aqueous phase was saturated with NaCl and the phases were separated. The aqueous phase was extracted with DCM (1.0 mL, 2.0v) twice, the organic phases were combined, and 50 mg of biphenyl in 2 mL of MeCN was added as an internal HPLC standard. Solution yield was assessed by HPLC. Reaction results are summarized in the following table:
Example 7: Base Screens for the Synthesis of 5,5-dimethyl-3-methylenepyrrolidin-2-one
[0074] In this experiment, various concentrations of NaOH were tested as described below:
[0075] 2,2,6,6-tetramethylpiperidin-4-one (500.0 mg, 3.06 mmol, 1.0 eq. (“starting material”), tetrabutylammonium hydroxide (0.12 g, 0.153 mmol, 0.050 eq), and chloroform (0.64 g, 0.4 mL, 5.36 mmol, 1.75 eq.) were charged into a vial equipped with a magnetic stir bar. The vial was cooled in an ice bath, and a solution of an amount wt% sodium hydroxide as shown in the Table below in water (0.98 g, 24.48 mmol, 8.0 eq.) was added drop wise over 2 min. The reaction mixture was stirred until completion and assessed by GC analysis. The reaction mixture was diluted with DCM (2.0 mL, 4.0v) and H2O (3.0 mL, 6.0v). The phases were separated and the aqueous phase is extracted with DCM (1.0 mL, 2.0v). The organic phases were combined and 2 M hydrochloric acid (0.17 g, 2.3 mL, 4.59 mmol, 1.5 eq.) was added. The reaction mixture was stirred until completion, assessed by HPLC. The aqueous phase was saturated with NaCl and the phases were separated. The aqueous phase was extracted with DCM (1.0 mL,
2.0v) twice, the organic phases were combined, and 50 mg of biphenyl in 2 mL of MeCN was added as an internal HPLC standard. Solution yield was assessed by HPLC.
Reaction results are summarized in the following table:
Example 8: Phase Transfer Catalyst (PTC) Synthesis of 5,5-dimethyl-3-methylenepyrrolidin-2-one
[0076] Various amounts of PTCs were tested as described below:
Tetrabutylammonium hydroxide (0.01 eq.), TBAB (0.01 eq.), Tributylmethylammonium chloride (0.01 eq.), Tetrabutylammonium hydroxide (0.02 eq.), TBAB (0.02 eq.), Tributylmethylammonium chloride (0.02 eq.), Tetrabutylammonium hydroxide (0.03 eq.), TBAB (0.03 eq.), Tributylmethylammonium chloride (0.03 eq.).
[0077] 2,2,6,6-tetramethylpiperidin-4-one (500.0 mg, 3.06 mmol, 1.0 eq. (“starting material”)), PTC (0.12 g, 0.153 mmol, 0.050 eq), and chloroform (1.75 eq.) were charged into a vial equipped with a magnetic stir bar. The vial was cooled in an ice bath, and a solution of 50 wt% sodium hydroxide (0.98 g, 24.48 mmol, 8.0 eq.) was added drop wise over 2 min. The reaction mixture was stirred until completion, assessed by GC analysis. The reaction mixture was diluted with DCM (2.0 mL, 4.0v) and H20 (3.0 mL, 6.0v). The phases were separated and the aqueous phase was extracted with DCM (1.0 mL, 2.0v). The organic phases were combined and 2 M hydrochloric acid (0.17 g, 2.3 mL, 4.59 mmol, 1.5 eq.) was added. The reaction mixture was stirred until completion, assessed by HPLC. The aqueous phase was saturated with NaCl and the phases were separated. The aqueous phase was extracted with DCM (1.0 mL, 2.0v) twice, the organic phases were combined, and 50 mg of biphenyl in 2 mL of MeCN was added as an internal HPLC standard. Solution yield was assessed by HPLC. The reaction results are summarized in the following table:
Reactions Conditions Result
8D Tetrabutylammonium hydroxide Almost complete
(0.02 eq.) overnight (2% starting
material), 82% solution yield
8E TBAB (0.02 eq.) Almost complete
overnight (2% starting material), 71% solution yield
8F Tributylmethylammonium chloride Incomplete overnight (4%
(0.02 eq.) starting material), 72%
solution yield
8G Tetrabutylammonium hydroxide Almost complete
(0.03 eq.) overnight (3% starting
material), 76% solution yield
8H TBAB (0.03 eq.) Almost complete
overnight (3% starting material), 76% solution yield
81 Tributylmethylammonium chloride Almost complete
(0.03 eq.) overnight (2% starting
material), 78% solution yield
Example 9. Preparation of 2,2,6,6-tetramethylpiperidin-4-one hydrochloride
2,2,6,6-tetramethylpiperidin-4-one 2,2,6,6-tetramethylpiperidin-4-one hydrochloride
[0078] 2,2,6,6-tetramethyl-4-piperidinone (30 g, 193.2 mmol, 1.0 eq) was charged to a 500 mL nitrogen purged three necked round bottomed flask equipped with condenser. IPA (300 mL, 10 vol) was added to the flask and the mixture heated to 60 °C until dissolved.
[0079] To the solution at 60 °C was added 5-6 M HC1 in IPA (40 mL, 214.7 mmol, 1.1 eq) over 10 min and the resulting suspension stirred at 60 °C for 30 min then allowed to cool to ambient temperature. The suspension was stirred at ambient temperature overnight, then filtered under vacuum and washed with IPA (3 x 60 mL, 3 x 2 vol). The cream colored solid was dried on the filter under vacuum for 10 min.
[0080] The wet cake was charged to a 1 L nitrogen purged three necked round bottomed flask equipped with condenser. IPA (450 mL, 15 vol) was added to the flask and the suspension heated to 80 °C until dissolved. The mixture was allowed to cool slowly to ambient temperature over 3 h and the resulting suspension stirred overnight at ambient temperature.
[0081] The suspension was filtered under vacuum, washed with IPA (60 mL, 2 vol) and dried on the filter under vacuum for 30 min. The resulting product was dried in a vacuum oven at 40 °C over the weekend to give a white crystalline solid, 21.4 g, 64% yield.
Example 10. Synthesis of (S)-2,2,4-trimethylpyrrolidine hydrochloride from (S)-3,5,5-trimethyl-pyrrolidin-2-one
[0082] Each reactor was charged with (,S)-3,5,5-trimethyl-pyrrolidin-2-one in THF, H2, and the catalyst shown in the below table. The reactor was heated to 200 C and pressurized to 60 bar, and allowed to react for 12 hours. GC analysis showed that (S)-2,2,4-trimethylpyrrolidine was produced in the columns denoted by “+.”
[0083] A 2.5% solution of (,S)-3,5,5-trimethyl-pyrrolidin-2-one in THF was flowed at 0.05 mL/min into a packed bed reactor prepacked with 2% Pt-0.5%>Sn/SiO2catalyst immobilized on silica gel. H2 gas was also flowed into the packed bed reactor at 20 mL/min. The reaction was carried out at 130 °C under 80 bar pressure with a WHSV (Weigh Hourly Space Velocity) of 0.01-0.02 h“1. The product feed was collected in a batch tank and converted to (S)-2,2,4-trimethylpyrrolidine HC1 in batch mode: 36%>
Hydrochloric acid (1.1 eq.) was added keeping the temperature below 20 °C. Distillation of the solvent, backfilling with isopropyl acetate (4v), was carried out to leave a residual volume of 5v. Karl Fischer analysis < 0.2% w/w H2O. MTBE (methyl tertiary butyl ether) (lv) was added at 20-30 °C and the solids were filtered off under nitrogen at 15-20 °C, washing with isopropyl acetate (1.5v) and drying under vacuum at 40-45 °C to give (S)-2,2,4-trimethylpyrrolidine hydrochloride as a white crystalline solid (74.8%> yield, 96.1% ee).
Alternate synthesis
[0084] A 2.5%) solution of (,S)-3,5,5-trimethyl-pyrrolidin-2-one in THF was flowed at 0.05 mL/min into a packed bed reactor prepacked with 4% Pt-2%>Sn/Ti02catalyst immobilized on silica gel. H2 gas was also flowed into the packed bed reactor at 20 mL/min. The reaction was carried out at 200 °C under 50 bar pressure with a WHSV (Weigh Hourly Space Velocity) of 0.01-0.02 h“1. The product feed was collected in a batch tank and converted to (S)-2,2,4-trimethylpyrrolidine HC1 in batch mode: 36%
Hydrochloric acid (1.1 eq.) was added keeping the temperature below 20 °C. Distillation of the solvent, backfilling with isopropyl acetate (4v), was carried out to leave a residual volume of 5v. Karl Fischer analysis < 0.2% w/w H2O. MTBE (methyl tertiary butyl ether) (lv) was added at 20-30 °C and the solids were filtered off under nitrogen at 15-20 °C, washing with isopropyl acetate (1.5v) and drying under vacuum at 40-45 °C to give (S)-2,2,4-trimethylpyrrolidine hydrochloride as a white crystalline solid (88.5% yield, 29.6%> ee).
Alternate synthesis
[0085] A 2.5% solution of (,S)-3,5,5-trimethyl-pyrrolidin-2-one in THF was flowed at 0.05 mL/min into a packed bed reactor prepacked with 2% Pt-0.5%>Sn/TiO2 catalyst immobilized on silica gel. H2 gas was also flowed into the packed bed reactor at 20 mL/min. The reaction was carried out at 150 °C under 50 bar pressure with a WHSV (Weigh Hourly Space Velocity) of 0.01-0.02 h“1. The product feed was collected in a batch tank and converted to (S)-2,2,4-trimethylpyrrolidine HC1 in batch mode: 36%>
Hydrochloric acid (1.1 eq.) was added keeping the temperature below 20 °C. Distillation of the solvent, backfilling with isopropyl acetate (4v), was carried out to leave a residual volume of 5v. Karl Fischer analysis < 0.2% w/w H20. MTBE (methyl tertiary butyl ether) (lv) was added at 20-30 °C and the solids were filtered off under nitrogen at 15-20 °C, washing with isopropyl acetate (1.5v) and drying under vacuum at 40-45 °C to give (S)-2,2,4-trimethylpyrrolidine hydrochloride as a white crystalline solid (90.9% yield, 98.0%> ee).
Alternate synthesis
[0086] A 2.5%) solution of (,S)-3,5,5-trimethyl-pyrrolidin-2-one in THF was flowed at 0.03 mL/min into a packed bed reactor prepacked with 2% Pt-8%>Sn/Ti02catalyst immobilized on silica gel. H2 gas was also flowed into the packed bed reactor at 40 mL/min. The reaction was carried out at 180 °C under 55 bar pressure with a residence time of 6 min. The product feed was collected in a batch tank and converted to (S)-2,2,4-trimethylpyrrolidine HC1 in batch mode: 36% Hydrochloric acid (1.1 eq.) was added keeping the temperature below 20 °C. Distillation of the solvent, backfilling with isopropyl acetate (4v), was carried out to leave a residual volume of 5v. Karl Fischer analysis < 0.2% w/w H2O. MTBE (methyl tertiary butyl ether) (lv) was added at 20-30 °C and the solids were filtered off under nitrogen at 15-20 °C, washing with isopropyl acetate (1.5v) and drying under vacuum at 40-45 °C to give (,S)-2,2,4-trimethylpyrrolidine hydrochloride as a white crystalline solid (90.4%> yield, 96.8%> ee).
Patent
WO 2019010092
PATENT
US 20160095858
https://patents.google.com/patent/US20160095858A1/en
Cystic fibrosis (CF) is a recessive genetic disease that affects approximately 30,000 children and adults in the United States and approximately 30,000 children and adults in Europe. Despite progress in the treatment of CF, there is no cure.
In patients with CF, mutations in CFTR endogenously expressed in respiratory epithelia leads to reduced apical anion secretion causing an imbalance in ion and fluid transport. The resulting decrease in anion transport contributes to enhanced mucus accumulation in the lung and the accompanying microbial infections that ultimately cause death in CF patients. In addition to respiratory disease, CF patients typically suffer from gastrointestinal problems and pancreatic insufficiency that, if left untreated, results in death. In addition, the majority of males with cystic fibrosis are infertile and fertility is decreased among females with cystic fibrosis. In contrast to the severe effects of two copies of the CF associated gene, individuals with a single copy of the CF associated gene exhibit increased resistance to cholera and to dehydration resulting from diarrhea—perhaps explaining the relatively high frequency of the CF gene within the population.
Sequence analysis of the CFTR gene of CF chromosomes has revealed a variety of disease causing mutations (Cutting, G. R. et al. (1990) Nature 346:366-369; Dean, M. et al. (1990) Cell 61:863:870; and Kerem, B-S. et al. (1989) Science 245:1073-1080; Kerem, B-S et al. (1990) Proc. Natl. Acad. Sci. USA 87:8447-8451). To date, greater than 1000 disease causing mutations in the CF gene have been identified (http://cftr2.org). The most prevalent mutation is a deletion of phenylalanine at position 508 of the CFTR amino acid sequence, and is commonly referred to as F508del. This mutation occurs in approximately 70% of the cases of cystic fibrosis and is associated with a severe disease.
The deletion of residue 508 in F508del prevents the nascent protein from folding correctly. This results in the inability of the mutant protein to exit the ER, and traffic to the plasma membrane. As a result, the number of channels present in the membrane is far less than observed in cells expressing wild-type CFTR. In addition to impaired trafficking, the mutation results in defective channel gating. Together, the reduced number of channels in the membrane and the defective gating lead to reduced anion transport across epithelia leading to defective ion and fluid transport. (Quinton, P. M. (1990), FASEB J. 4: 2709-2727). Studies have shown, however, that the reduced numbers of F508del in the membrane are functional, albeit less than wild-type CFTR. (Dalemans et al. (1991), Nature Lond. 354: 526-528; Denning et al., supra; Pasyk and Foskett (1995), J. Cell. Biochem. 270: 12347-50). In addition to F508del, other disease causing mutations in CFTR that result in defective trafficking, synthesis, and/or channel gating could be up- or down-regulated to alter anion secretion and modify disease progression and/or severity.
Accordingly, there is a need for novel treatments of CFTR mediated diseases.

////////////////OLACAFTOR, VX 440, Phase II, Cystic fibrosis, CS-0044588, UNII-RZ7027HK8F, RZ7027HK8F
CC1CC(N(C1)C2=C(C=CC(=N2)C3=CC(=CC(=C3)F)OCC(C)C)C(=O)NS(=O)(=O)C4=CC=CC=C4)(C)C

